Chemical principles (Chemotherapy)
Contents
We sketch the predominant working mechanisms of the three main types of cytotoxic agents used in the treatment of head and neck cancers: DNA cross linking agents, antimetabolites and protein inhibitors. All of these and additional mechanisms are known in great detail and are the subject of ongoing research. We refer to the ample scientific and medical literature for detailed descriptions and ongoing discussions of chemotherapy drugs and their properties. Please keep in mind that chemotherapy plays a less prominent role in the treatment of head and neck cancers than for other malignancies.
The following merely gives a coarse picture, an impression of the main underlying biochemical and physiological principles that make these various agents cytotoxic and thus suitable as chemotherapy drugs. As will be seen, none of these three types of chemotherapy drugs are specifically targeting malignant cells but exploit general properties of living and dividing cells, often causing considerable adverse side effects. Accordingly, much contemporary research about chemotherapy drugs is focussed on finding and testing substances that act more specifically on malignant cells and cause less damage to healthy cells. This research is moving in a more biologically and biochemically informed direction than traditional chemotherapy research over several past decades.
DNA crosslinking agents
Since the accidental discovery in 1965 by Rosenberg at el. that inorganic compounds such as cisplatin inhibit cell division, this compound has been widely used in chemotherapy. It is an unusual pharmaceutical in that almost all other medicinal drugs are organic compounds. Cisplatin is a highly potent (and highly generally toxic) agent against a number of malignant tumours.
Cisplatin, cis-(NH3)2PtCl2, is usually administered intravenously in saline solution. Activation of the cisplatin complex occurs in the intracellular environment where the chlorine concentration is notably lower than that of the extracellular space: the chlorine ligands are replaced by weakly bonded water molecules, increasing the reactivity of the cis-(NH3)2Pt fragment (see Figure 1). The increased potency of cisplatin in this activated form results in its reaction with various molecular cell targets. Though various potential reactive sites exist within a cell, interaction with the DNA is considered to be the primary target. There, the predominant interaction is the formation of 1,2-intrastrand cross links between adjacent purine bases. The activation of cisplatin, followed by binding of the cis-(NH3)2Pt fragment to a preferred site in a particular groove in the DNA strand is shown in Figure 1: the fragment fits this groove particularly well.
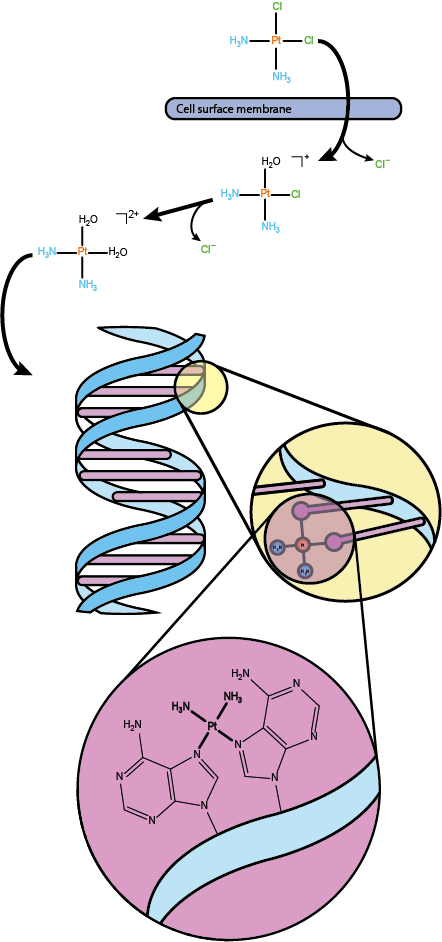
Such interaction induces a conformational change to the structure of the DNA causing it to both bend and unwind to varying degrees. Since DNA replication is at the heart of all cell replication, any hindrance of DNA and its replication will inhibit cell division and replication in some way.
Though cisplatin exposure is clearly linked to disruption in the replication and transcription processes of the cell, such effects do not necessarily and immediately result in cell death. Instead pro-apoptotic signals triggered in the cell in response to DNA damage can also activate various DNA repair mechanisms. DNA damage caused by cisplatin tends to be poorly repaired in contrast to that caused by adducts with less cytotoxic molecules. Effective DNA repair mechanisms are obviously extremely useful for the successful replication of healthy cells to avoid errors in DNA and cell replication (mutations). However, occasionally tumour cells exhibit or develop resistance to cisplatin. In such cases an enhanced nucleotide excision repair (NER) pathway, the mechanism responsible for DNA repair, has been observed alongside further resistance mechanisms such as reduced drug uptake or increased drug inactivation or increased excretion of the drug molecules from the cell.
Like most other water-soluble metabolites, cisplatin is eliminated from the body via the kidneys, leading to an accumulation of the drug in the kidneys. While other antineoplastic drugs causing DNA damage (see below) predominantly do harm to rapidly dividing cells, cisplatin causes considerable damage also to some more slowly dividing cells such as those in renal tubules by a number of different mechanisms. In fact, the nephrotoxicity of cisplatin is a significant treatment-limiting factor.
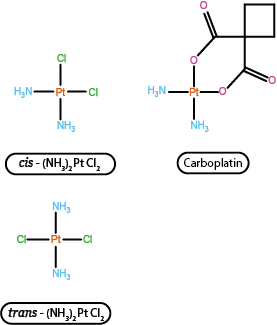
In some cases, carboplatin may be used instead. Carboplatin exhibits greater stability with regard to hydrolysis and thus tends to cause less renal damage because predominantly a less aggressive version of the drug molecule is excreted via the kidneys. The increased stability to hydrolysis arises as a function of the different leaving group in the molecule, replacing the chlorine ions in cisplatin by an organic molecule fragment. The overall cytotoxic properties of carboplatin are otherwise largely very similar to those of cisplatin. Figure 2 highlights the similarities and differences between the two platinum antineoplastic drugs.
Cisplatin and carboplatin are not the only substances interfering with DNA replication by interacting with the DNA strands in some way. For example, an organic compound called bleomycin is also used in chemotherapy and is thought to hinder DNA replication by binding to it. Bleomycin has been known for a long time and after its discovery was originally intended as an antibiotic but was found to be too toxic for that purpose.
Antimetabolites
A group of drugs commonly known as antimetabolites also aim to cause damage to DNA, though in ways very different from the damage caused by cisplatin. Antimetabolites mainly harm rapidly dividing cells. They are structurally similar to normal metabolites yet display small but crucial chemical differences. Antimetabolites inhibit cellular processes by essentially taking part in them and competing with the normal metabolites, consequently inducing cell death by blocking metabolic pathways or leading to inviable faulty products in the cell cycle.
The two commonly used antimetabolites used in the treatment of head and neck cancers are 5-fluorouracil and methotrexate. The two substances are shown together with their respective normal metabolite counterparts in Figure 3.
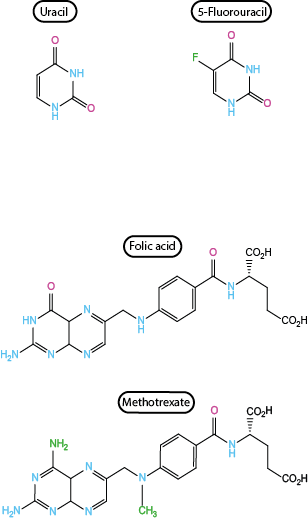
Thymidine (T) is one of the four building blocks of DNA. 5-Fluorouracil and methotrexate belong to the same type of antimetabolite (further target metabolites exist and are being used for the treatment of other malignancies) and both ultimately inhibit the thymidine biosynthesis pathways. Cells which are rapidly synthesising DNA, such as malignant cells undergoing frequent replication, will therefore experience a relatively more severe depletion of thymidine (and consequently cell death) than slowly replicating cells. A further common feature of these two antimetabolites is their pursuit of a variety of metabolic mechanisms. For example, methotrexate in low dosage is also used in the treatment of severe cases of arthritis or Crohn’s disease as one of its effects is immunosuppression, in addition to being cytotoxic.
5-Fluorouracil
As an analogue of uracil (U), a pyrimidine base utilised in RNA and as a precursor to thymidine synthesis, 5-fluorouracil (5-FU) is a potent antimetabolite. 5-FU is a prodrug (an inactive compound that is metabolised to produce a therapeutic agent) and its modes of action depend on which of the various metabolic pathways are followed after administration of the prodrug. Fundamentally all these routes result in 5-FU being activated to one of multiple nucleotide forms and going on to cause damage to DNA and/or RNA. RNA is a single-stranded polymeric chain, normally folded onto itself, while DNA is a double-stranded helix. RNA is required for genetic information to be read from DNA and translated in the process of cell division and in the production of specific proteins. A whole range of RNA versions with slightly different roles in these processes are known.
One course of events in which 5-FU prevents the synthesis of thymidine is summarised in Figure 4.
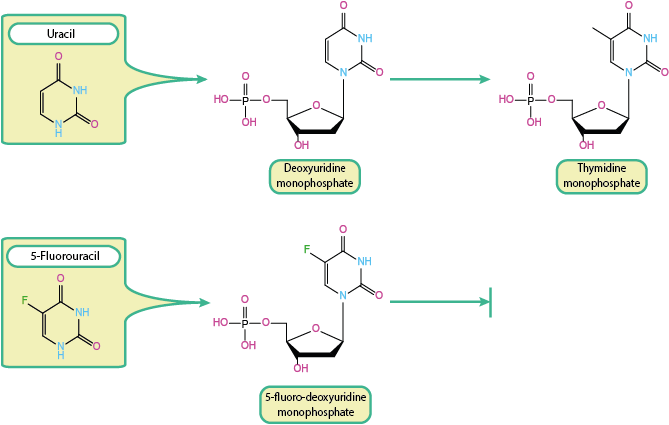
Thymidine (T) is an essential component of DNA, produced by the methylation of deoxyuridine. This reaction is as catalysed by an enzyme, thymidylate synthase. Deoxyuridine, in turn, is derived from uracil. This normal course of events is shown at the top of Figure 4. With 5-FU being a close analogue of uracil it is unsurprising that, once activated, 5-FU is able to interfere with the synthesis of thymidine. This is shown in the bottom trace of Figure 4. Taking part in the expected biosynthesis route, 5-FU is converted to the fluorine analogue of deoxyuridine monophosphate. At this stage it forms a strong covalent link with the enzyme that normally catalyses the next reaction step (thymidylate synthase), production of thymidine monophosphate. The presence of the fluorine atom in 5-FU in the place of a hydrogen atom in normal uracil results in a permanent binding with the enzyme, rendering the enzyme useless and thymidine synthesis is halted. This impairs DNA synthesis due to lack of thymidine supply and eventually results in so-called thymineless cell death.
This metabolic pathway of 5-FU is shown in Figure 5, together with two further pathways that allow 5-FU to interfere with the life cycle of a cell.
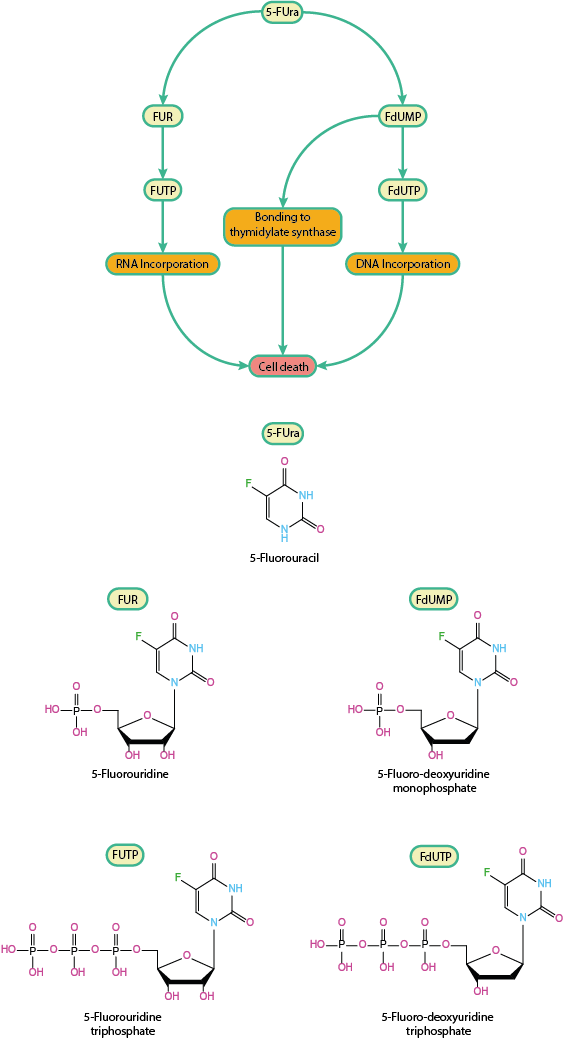
The pathway illustrated in Figure 4 (and included in the map in the top part of Figure 5) is causing cell death by preventing the production of a necessary building block of DNA. When 5-FU is incorporated in a different branch of the metabolism of a cell, it disrupts RNA synthesis (see left branch of map in Figure 5): metabolic conversion of 5-FU to 5-fluoro-uridine triphosphate (FUTP) via 5-fluorouridine (FUR) provides an alternative substrate for RNA polymerase. RNA polymerase is an enzyme that is essential in the construction of the polymeric RNA chains (RNA in turn is an essential element of all living cells; it is needed to copy and translate information from DNA). Consequently, FUTP is incorporated into cellular RNA where it is then able to adversely affect all kinds of metabolic processes involving RNA.
Similarly, when 5-FU is converted to the deoxyuridine equivalent (FdUTP) it can provide an alternative substrate to DNA polymerase, with similar disruptive effects on DNA synthesis in the cell. As yet another possible result of 5-FU metabolism, FdUTP may be incorporated into DNA to produce similar disruptive effects caused by the faulty DNA version (symbolised by the right branch in the map, Figure 5). This is a less common effect owing to the action of enzymes that ordinarily prevent uracil incorporation into DNA.
The general pattern remains throughout that very subtle, minor changes in molecular structure (see, for example, bottom part of Figure 5) have drastic and varied effects on the cell metabolism.
Methotrexate
Belonging to a class of antimetabolites known as antifols, methotrexate (MTX) is a close analogue of folic acid (see Figure 3). Folic acid plays a key role in thymidine synthesis. Folic acid and its derivatives belong to the family of B vitamins. MTX, by virtue of this similarity to folic acid is able to interfere with the folate metabolism through multiple pathways. The mechanism by which MTX hinders thymidine production in the cell is very different from that by which 5-FU interferes with thymidine synthesis. This may be taken as an illustration of the multiple metabolic cellular pathways in the cell cycle, offering both multiple opportunities for targeted interventions and chances for unwanted (and sometimes unforeseen) effects.
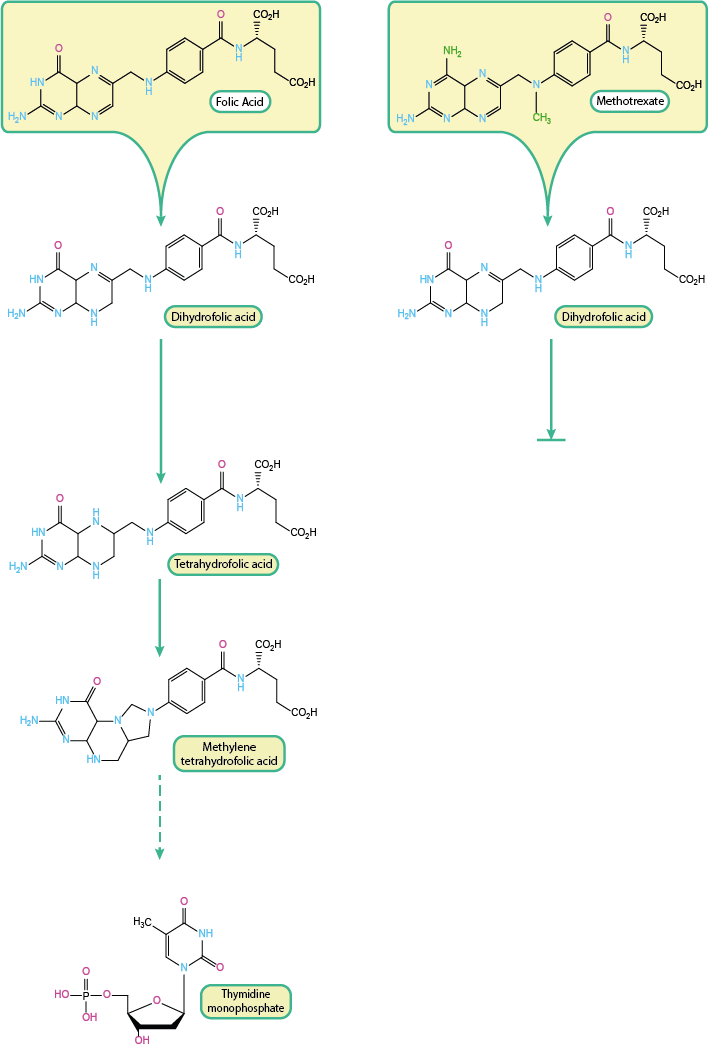
The left part of Figure 6 illustrates the stepwise regular metabolic transformation of folic acid into methylene tetrahydrofolic acid, the compound which acts as the source of the carbon atom / methyl group in thymidine. Each of these steps is catalysed by enzymes. The enzyme dihydrofolate reductase (DHFR) is responsible for enabling the step from dihydrofolic acid to tetrahydrofolic acid.
Competitive inhibition of DHFR by MTX prevents the conversion of dihydrofolic acid to tetrahydrofolic acid as part of the biosynthesis of thymidine (Figure 6, right). The affinity of MTX to this receptor is about a factor 103 higher than that for the normal substrate. The depletion of tetrahydrofolic acid in turn creates a deficiency of methylene tetrahydrofolic acid, which is involved in the methylation of deoxyuridine to thymidine. Therefore the resulting lack of thymidine results in a decrease in DNA synthesis and consequently cell replication is inhibited.
Folate growth factors are known to assume polyglutamate (polymeric) forms within the cell which are a preferred substrate of DHFR. MTX also has the potential to be converted into polyglutamate derivatives with a higher affinity for DHFR in comparison to MTX itself. The MTX polyglutamate is able to accumulate within the cell and achieves a more prolonged suppression of DHFR. The higher affinity of MTX polyglutamate and its longer intracellular half-life makes this mode of action more effective.
As a consequence of the inhibition of DHFR oxidised folates are produced and accumulate in the cell. These oxidised folates have been shown to have potential to inhibit a variety of enzymes associated with purine synthesis, another important metabolic pathway especially for the synthesis of adenine and guanine (two further of the four basic building blocks of DNA). Such enzymes may also be inhibited by the MTX polyglutamate forms.
Only a small quantity of the DHFR enzyme is required to maintain normal metabolic activity in the cell. The regulation of the cell metabolism responds to lower concentrations of tetrahydrofolic acid (see left part of Figure 6) by producing more DHFR. Accordingly, higher DHFR concentrations in the cell can counteract MTX and are the underlying cause for the development of MTX resistance.
Protein inhibitors
The mode of action of the two types of antineoplatic drugs discussed so far is best summarised as cytotoxic, that is killing cells by interfering with basic building blocks and reactions in the cell metabolism. The mode of action of so-called protein inhibitors is better called cytostatic, that is inhibiting cell growth. Because of this difference in mode of action there were early hopes that such drugs would have less severe side effects than conventional chemotherapy drugs. That turns out to be not the case.
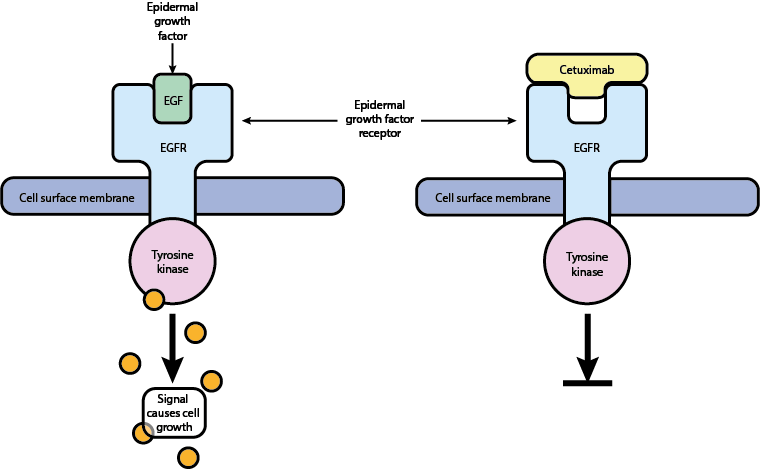
Figure 7 illustrates the working principle of one such protein inhibitor, cetuximab. Epidermal growth factor (EGF) and its complimentary receptor (EGFR) ordinarily orchestrate the growth of cells. The binding of EGF to its receptor results in the affected cell being permitted to multiply which, in a healthy mature system, is only rarely required. Mutations leading to upregulation of these proteins culminate in the proliferation of malignant cells by bypassing this strict growth control mechanism. The ability of malignant cells to produce their own EGF allows continuous and rapid growth.
Through competition with endogenous ligands that bind to the EGFR, cetuximab acts to inhibit the stimulation of this receptor by blocking it. In this way cetuximab prevents phosphorylation of the enzyme tyrosine kinase associated with EGFR, resulting in no further cell signalling and no further cell growth.
Obviously, only tumour lines expressing particular growth factors / receptors will be susceptible to this kind of ‘biological’ therapy (a well known case from public dispute about NHS support is the treatment of some breast cancers with trastuzumab). Cetuximab belongs to a class of biological macromolecules (very large molecules) called monoclonal antibodies. These are so large that they cannot easily (or not at all) transgress the cell membrane and are thus limited to acting on receptors and structures on the cell surface, such as EGFR.
Another mechanism to hinder the process of cell division and growth is to interfere with the late stage of cell division and the assembly of daughter cells.
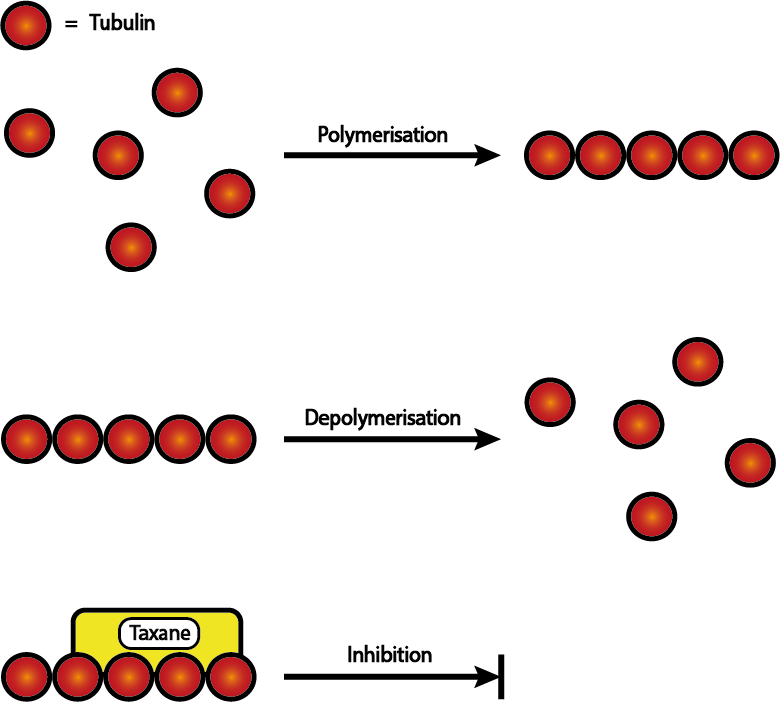
Figure 8 illustrates the working principles of the late stages of cell division (mitosis) and how taxanes interfere with this process. Mitosis (the late stage of cell division) requires mitotic spindles to form to provide structure and to separate genetic material between the two resulting daughter cells in an orderly and controlled fashion. Polymerisation of smaller components (tubulin) allow these spindles to form (Figure 8 top) and to retract in the next stage of separation into two daughter cells, with the relevant genetic material attached, by depolymerisation (Figure 8 middle). Both of these steps are essential for successful cell replication and thus offer another target for antineoplastic therapies.
Taxanes are specifically concerned with the depolymerisation process (Figure 8 bottom). By stabilising the polymeric microtubules, taxanes prevent the mitotic spindles from retracting. This restriction prevents the continuation of mitosis and consequently the successful formation of daughter cells.