Delivery methods
Contents
Below we discuss in slightly more detail the general principles of the journey of a medicinal drug through the body. We also sketch some basic principles and properties of organic molecules in the solid state, how these properties impact the preparation and suitability / bioavailability of drugs, how and why the production conditions are crucially important for quality control – and how this all relates to instructions on medication packs on how to store and use the medication.
Drug timings and dosage
Drug activation and deactivation
Once a drug has been absorbed, has reached its site of action and performed its function, it has to be eliminated from the body.
For systemically delivered drugs, the bloodstream delivers the drug to all of the body’s organs. The distribution of the drug between the organs depends on their size and blood supply. Drugs carried in the bloodstream are present in two forms, either bound to proteins in the blood or unbound. For a drug to be effective it has to be predominantly unbound: only in this form will it be able to pass through membranes and reach its site of action. The bloodstream also takes the drug to those organs that will eliminate it from the body. The main removal organs are the liver where most drugs are metabolised (broken down or otherwise chemically modified) and the kidneys from where drugs and/or their metabolites are excreted into urine.
The kidneys can excrete water-soluble substances. These are substances that have a low molecular weight and/or are polar, or even fully ionised, molecules at physiological conditions (pH; the acidity levels of body fluids). Most drug molecules do not belong to this category of water-soluble substances, most drugs are lipophilic (soluble in fat) organic substances. In the liver, these drug molecules are processed (metabolised) and chemically modified to transform them into compounds that are water-soluble and can be readily excreted via the kidneys. This process is illustrated in Figure 1.

For some drugs, the molecules are broken down into smaller, more readily water-soluble fragments. Alternatively, drug molecules can be chemically modified (usually by attaching polar groups, such as OH groups to the molecular frame) to obtain more readily soluble products. Most products of this process are less active than the parent compound. In some cases, however, metabolites may be responsible for toxic, mutagenic, teratogenic or carcinogenic effects.
For example, the analgesic ibuprofen is a non-polar organic substance that is insufficiently soluble in water for direct excretion by the kidneys. Instead, the drug is almost completely metabolised in the body before excretion. The primary route of elimination for ibuprofen is by metabolism in the liver (using the cytochrome P450 enzymes; the main chemical ‘factory’ in the liver for the processing of drug molecules and other substances) where the ibuprofen molecules are transformed into inactive but more soluble metabolites, carboxy-ibuprofen and 2-hydroxy-ibuprofen. These two metabolites are soluble in water, so can be excreted via the kidneys.
Sometimes metabolites are actually the active therapeutic substance. In this case, the drug being taken is called a pro-drug. A pro-drug is a substance that has no, or a lower, activity against a target site than one of its major metabolites. A well-known example of a pro-drug is codeine which needs to be metabolised to its active metabolite morphine. The delivery of pro-drugs is symbolised in Figure 2.
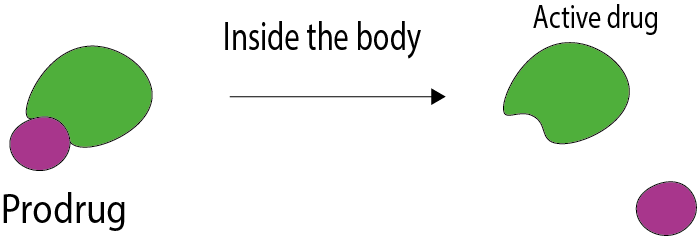
In general, pro-drugs are used to improve delivery or kinetic properties of a drug. Use of a pro-drug is sometimes a way to reduce toxicity of a drug, or to achieve a more target-specific delivery (to specific cells or tissues).
An important example of a pro-drug is levodopa, which is used to treat Parkinson’s disease (a degenerative neurological condition where lack of dopamine production in the brain leads to parts of the brain not functioning properly). Levodopa is an amino acid precursor to dopamine. Dopamine cannot be directly replenished in the brain of patients with Parkinson’s disease because the dopamine molecules are too polar to be able to cross the blood-brain barrier. While levodopa is also a polar molecule, it does get complexed by proteins that carry amino acids across the blood-brain barrier. Once in the brain, levodopa is converted to dopamine by the enzyme DOPA decarboxylase.
For a drug to reach its site of action and to produce a response, it must first pass through multiple membranes in the body. Figure 3 shows a sketch of the structure of a membrane.
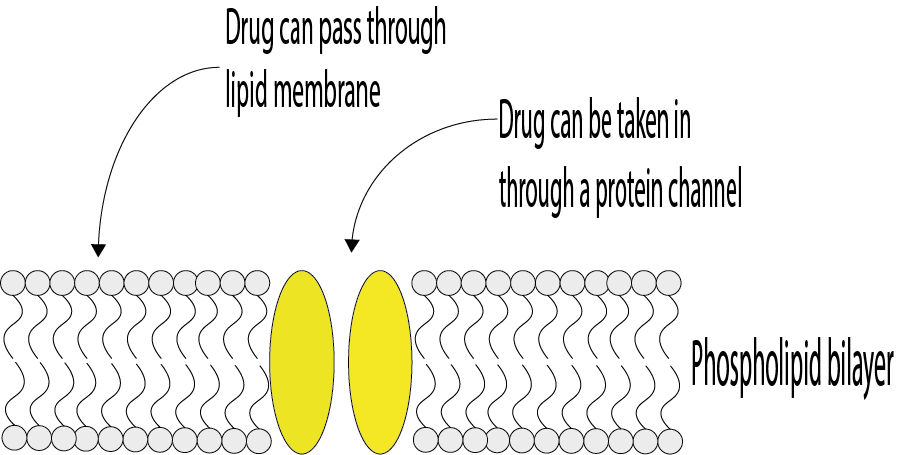
The solubility of a drug in both aqueous and lipid (fatty, non-polar) environments will determine how easily it can pass through membranes because membranes are made of lipids. To pass through the body’s membranes a drug needs to either be soluble in lipids, or it needs to mimic some of the body’s natural compounds (like amino acids) to pass through protein channels in the membrane (see Figure 3). How well a drug can pass through membranes and travel around the body will have an effect on how a drug has to be administered. The target site, and the membranes the drug must pass through to get there, will have an effect on the acceptable size of a drug molecule. For example, drugs that need to get through the blood-brain barrier will need to be smaller than for passage through most other membranes.
There are several types of components that drugs interact with in order to produce physiological and biochemical effects. The most common sites of interaction are proteins (receptors and enzymes) and DNA. When a drug interacts with an enzyme it can act as an inducer (increasing enzyme synthesis) or as an activator (increasing enzyme activity). Some drugs act as inhibitors (decreasing enzyme activity). Drugs that interact with receptors can either act as agonists (increasing the receptor response) or as antagonists (decreasing the desired response). Some drugs may act as an antagonist in one part of the body but as an agonist in another part.
Kinetics of drug delivery and elimination
When taking a single dose of a drug orally, its concentration will initially increase and then decrease over time. This behaviour is shown in the graph in Figure 4.
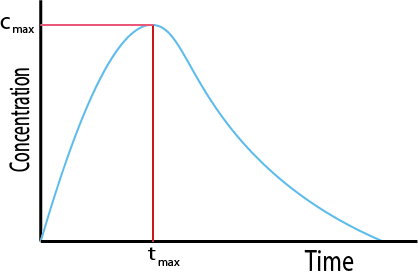
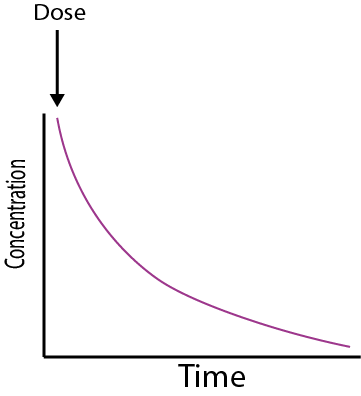
Cmax is the maximum concentration the drug achieves in the body. It achieves this concentration at a time tmax. Cmax and tmax depend on how quickly the drug enters the body, and then how quickly it is eliminated. The total area under the curve is a measure of the total systemic exposure to the drug. The curve of the graph before the peak gives information about how quickly the drug enters the systemic circulation. The area under the curve up to Cmax is therefore a measure of early exposure to the drug in the body. The graph in Figure 4 shows the change in concentration with time typical for an orally administered drug. Other methods of administration will have different concentration profiles. For example, for intravenous administration the drug is delivered directly to the bloodstream, and so the concentration of the drug starts at the maximum and then decays over time (see Figure 5).
Different drugs have different kinetics of elimination. They can either be eliminated by a first order process (see Figure 6, left graph) or by a zero order process (see Figure 6, right graph).

For most drugs and most methods of administration, elimination from the body follows first order kinetics. This means that a constant fraction of the drug present will be eliminated in a unit of time. Thus, the concentration of the drug in the body decreases logarithmically over time: the rate of elimination is proportional to the drug concentration, and the amount of drug removed per unit time will vary in direct proportion to the drug concentration. Most drugs follow first order elimination kinetics because generally therapeutic drug concentrations are not sufficiently high to saturate (one of the steps in) the elimination mechanism. Only once the elimination mechanism is saturated will zero order kinetics apply.
For an elimination process according to zero order kinetics, the concentration of the drug in the body decreases linearly over time: the rate of elimination is independent of the drug concentration, a constant amount of the drug is eliminated in a unit of time.
An example of a zero order elimination drug is ethanol (alcohol). The first metabolising step in the alcohol elimination process is oxidation to acetaldehyde and the corresponding enzyme is rapidly saturated, even at low alcohol concentrations. It is straightforward to calculate the amount of alcohol in the blood, based on this zero order elimination process. Alcohol breathalysers exploit these kinetic data. As the blood travels around the body, it picks up alcohol that you have been drinking. When the blood then goes into the lungs, a small representative proportion of alcohol gets passed from the blood into the lungs via gaseous exchange (similar to carbon dioxide in normal breathing). The concentration of alcohol in the blood that evaporates into breath directly depends on the concentration of alcohol in the body. The blood to breath ratio of alcohol is not constant, varying both with the individual and over time. However, it remains constant enough for the British Government to use a blood to breath ratio of 2,300 : 1. This means that the concentration of alcohol in the blood is 2300 times that of the concentration in the breath. This small proportion of alcohol in breath, together with some of these individual differences, are the reason for compulsory blood alcohol tests when you are caught driving under the influence of alcohol. The breathalyser test alone is not sufficiently accurate to determine exact alcohol levels in the body.
Figure 7 (first order kinetics) and Figure 8 (zero order kinetics) show how the rate of elimination can be expressed in terms of the half-life (t1/2) of the drug. The half-life is the time taken for 50% of the drug to be eliminated from the body.
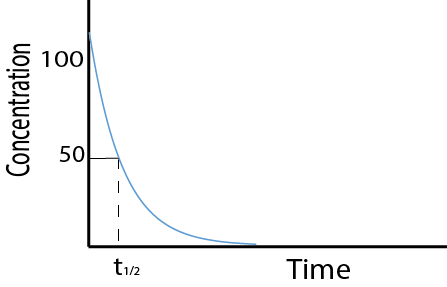
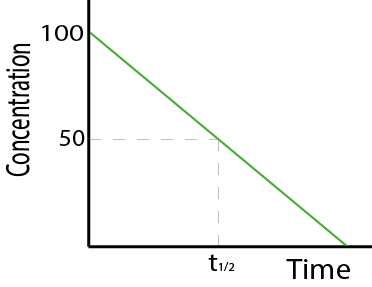
Alternatively, the elimination kinetics of a drug can be expressed as a rate constant, k. The rate constant is the fraction of the drug eliminated per time unit. Zero or first order characteristics of drug elimination processes have large effects on t1/2 and k.
For some drugs, the elimination kinetics are more complicated and are dose / concentration dependent. This means that as the concentration increases, the half-life of the drug will also increase. This will result in the concentration of the drug increasing disproportionally with increases in dose. This, in turn will eventually result in the elimination rate becoming independent of the concentration.
For drugs that are rapidly absorbed, a short t1/2 may cause the concentration to fall below the minimum effective concentration between doses unless the dose is large or the intervals are short. If the dose is large, the peak concentration may exceed the maximum safe concentration. Avoidance of toxicity and maintenance of a steady effect are easier to achieve with drugs for which t1/2 is relatively long. Sometimes it is desirable to attain a particular drug effect rapidly. This may make it necessary to give a so called loading dose. A loading dose is larger than the usual maintenance dose.
Drug Variability
The probability of being able to achieve effective therapy without the patient experiencing harmful effects is a measure of the drugs therapeutic utility. For a drug to be therapeutically useful, this probability must ideally be in excess of 50 %. For an individual drug, a decision must be made as to what extent the beneficial effects of the drug outweigh its adverse effect. The decision will be very dependent on both the drug and the condition being treated.
For different people taking the same drug there can be significant variability in both the desired and adverse effects. For the same drug people will have therapeutic windows of different locations and widths. The therapeutic index of a drug reflects a ratio of the blood concentration at which the drug is toxic, and the concentration at which the drug is effective. To be safe, drugs need as high a therapeutic index as possible. Different reactions of different people to the same drug can lead to some people having either an intolerance or an allergic reaction to a drug that other people are fine with. Whether a person is intolerant, allergic or has no response to a drug depends on the person’s genetics.
Variability between people can occur in both the drugs pharmacokinetics and the drugs pharmacodynamics, with the magnitude and relative contribution of each being dependent on the drug. There are many reasons that can explain why different people will respond differently to the same drug, including age, body weight, disease, comorbidities and other drugs that are simultaneously being taken. Generally the average dosing regimen will need to be modified to whether it is being given to a child, an adult or a senior. Gender can have some effect on variability, but generally this effect is very small (with the noticeable exception of alcohol).
Another major reason for variability is related to inheritance and genetics. However, this is highly unpredictable, especially for the pharmacodynamics involved. Reactions due to genetic disposition however will remain the same for a person’s lifetime. Differences in a person’s genetic structure can affect the way their body will absorb, distribute, metabolise and excrete a drug.
Differences can also result from an individual completely lacking a target receptor, causing the person to completely fail to respond to a drug. The sensitivity of a receptor to a drug can also vary between people, resulting in different drug responses.
The study of the effect that genetics has on drug response is called pharmacogenetics. This branch of pharmacological research is increasingly important, with more and more biologically derived and ‘personalised’ medications being developed.
Different medicinal drug preparations
Before any of the considerations about dosage and timings of drugs, different delivery modes, therapeutic windows, drug interactions and so on, are of any relevance, there is a long and hard journey for any ‘aspiring substance’ to become a useful and useable medicinal drug. Only very few candidate molecules make it from the designer stage in the laboratory (or more precisely: nowadays usually in a computer algorithm before ever entering a real laboratory) to any trial stages, let alone to the market in the form of a licensed drug.
Of course, there are all the obvious considerations that a substance must have the desired effect without too many serious side effects, it must be safe to produce and to store, it must be economically viable to make and distribute it; the list is long. Before any such practical considerations may play a role, there are a handful of fundamental properties that are crucial for a substance to be (or become) a useful drug, or not.
Irrespective of the mode of delivery of a drug (orally, or by injection) or its purpose, there is one essential property that determines its potential usefulness and bioavailability: its solubility. The majority of medicinal drugs are small to medium-sized organic molecules, the majority of these are solid at room and body temperature. For these molecules to reach their target site(s) in the body, they have to be soluble in water as well as in some less polar environments (such as fat and some other body fluids). If the compound is liquid under normal conditions, it still has to be miscible with the various relevant body fluids. In addition to the transport of drug molecules to their target site being dependent on their solubility properties, sufficient solubility at the target site is a requirement to achieve the necessary concentration of a drug at its site of action.
Solubility in different solvents is a fundamental property of each individual chemical substance. Solubility of medicinal drug compounds in water is so important for their function that medicinal drugs for oral use are commonly classified according to their solubility / permeability in the digestive system. Poor water solubility causes limited absorption, poor distribution in body tissues and poor metabolism characteristics.
Many organic substances in the solid state display a phenomenon called polymorphism: they tend to crystallise in different forms (see Figure 9 for an illustration) under ever so slightly different conditions.

The substance will be perfectly chemically pure (and chemically identical) in both crystalline forms, but because the two crystalline forms A and B have different geometric mutual arrangements (symmetries, to be precise), they will have different physical properties and only one of (sometimes many) the polymorphs will be thermodynamically stable. The other forms will, over time and given opportunity, eventually convert to this stable form. One of the physical properties depending on the crystalline form is solubility. Often, the most stable form is less soluble than some other polymorphs. However, some of the more soluble, less stable polymorphs may not be possible to maintain through the manufacture process, or may end up with a very short shelf life.
Preparing pure crystalline forms is a labour-intensive process and usually requires following a narrowly defined (and multiply tested) protocol. It involves all kinds of basic or sophisticated isolation and crystallisation techniques.
Figure 10 illustrates one of the possible production failures that may occur if not enough time is allowed for crystals to form, or if the growing conditions for crystallisation (concentration, temperature, purity of solvent, and so on) are not stable enough.

Under such circumstances a completely disordered, amorphous material may be obtained (Figure 10 top), or a so called partially ordered solid (powder) material is obtained (Figure 10 bottom; glasses belong to this category of partially ordered solid material). Partially ordered solids / glasses are sometimes thought of being a frozen snapshot version of the short- and medium-range order of a liquid. Again, these non-crystalline versions will have different properties from the crystalline varieties of the same chemical compound. Because of the lack of order, their properties will be fairly ill-defined and therefore, these forms are obviously unsuitable for medicinal drug use.
Another general trend displayed by many crystalline organic molecules is that their crystal structures are not particularly tightly packed, there may be spaces between molecules. This can have major implications for the suitability of a compound as a medicinal drug. Figure 11 illustrates this general property of many organic crystalline solids.

Empty spaces are there to be filled. From an energetic and structural point of view, it is attractive for many loosely held together solid-state structures of organic molecules to incorporate some solvent or water molecules. This may well increase the stability of the solid compound as these incorporated molecules help to hold the structure together. Another possibility is that solvent molecules might get trapped inside some hollow space of individual molecules and get encapsulated in that way. In either case, this is an unwanted effect as far as preparation of medicinal drugs is concerned. After dissolving the solid material, the trapped solvent molecules will be set free. If the trapped solvent molecules from the synthesis procedures happen to be toxic organic solvent molecules, this is obviously not wanted. One could argue that if the trapped solvent molecules are water molecules, it should not matter in terms of setting a toxic component free. While that is true, very rarely are these inclusions in any way controlled in terms of composition. That, again, leads to ill-defined and varying properties of different batches of preparation and rules out such compounds as suitable medicinal drug candidates.
Accordingly, pharmaceutical companies are obliged to follow tight regulatory processes and protocols to monitor their production lines. Having said all that, it is probably not surprising to hear that despite all these efforts, there are regularly reports about clinical failures of some (generic and otherwise) drug preparations. It takes very little to change the behaviour and properties of a medicinal drug – it could be very minor effects, such as a slightly different grain size of inert filler molecules to dilute and bulk up the active drug to pill size. Better understanding and property control of organic compounds in the solid state is therefore a prime research topic in all pharmaceutical industries. It is also fair to say that if you notice that the supposedly identical drug from company A does not work for you in the same way as the one manufactured by company B, this is likely not your imagination but a fact.
Given that organic molecules tend to be slightly temperamental and not as stable as a rock, some of the instructions you may find on the packaging of some of your prescribed (or over-the-counter) medication perhaps makes more sense:
- Best before dates for medicinal drugs (other than for supermarket-bought food) do make good sense: these are conservative estimates of periods of time over which it is highly unlikely that the drug preparation will have changed structurally.
- If it is required to keep a drug at low temperatures – this is either necessary to give the active form of a compound enough shelf life, or it is necessary because otherwise the active ingredient may chemically decompose.
- Most drugs are packaged such that they are protected from sunlight, in brown glass bottles or aluminium blister packs to exclude UV light. This is often necessary to prevent structural or chemical decomposition. Not only elevated temperatures do damage organic molecules, UV light can cause damage too (to organic molecules in pills just as to the organic molecules making up human bodies).
- Many drugs are packaged in individual one-pill compartments such that the pill only gets exposed to the air right before using it. Protection from moisture or oxygen in the air is often necessary to prevent decomposition of drug preparations (either the active ingredient or the filler compounds, or both).
- Some drugs are not pressed into a pill but are contained as powders in some kind of capsules. This might be necessary to prevent decomposition of a drug by stomach acid and to shuttle it safely to the intestines from where the active drug can then be absorbed by the body. Another reason might be that there is no way to press an active drug into a pill: applying pressure does alter the crystal structure of many organic molecules.
- If you purchase the same drug on the same day in two different forms as, say pills and in liquid form, you may notice that the quoted shelf life of the liquid (solution) preparation is typically shorter than that quoted for the solid preparation. This reflects another general property of organic compounds: they tend to be less stable once dissolved.
- Please dispose of unused drugs by returning them to your nearest pharmacy: many chemical substances used as medicinal drugs are harmful to the environment; your pharmacy will properly dispose of them.