Delivery methods
Understanding the effects of timing, delivery methods and interactions with your medication(s) not only helps to manage the routines of application, it makes it easier to spot signs and symptoms of unwanted medication-related effects.
Drug timings and dosage
When trying to understand a drug's effect, two main areas must be considered: the first is how the drug works and the effects it will have on the body (pharmacodynamics), the second is how your body will affect the drug and how the drug moves around the body (pharmacokinetics).
An important aspect of drug design, that will help determine the concentration of the dose given, is knowing how much of a given dose will reach its target site. Orally administered drugs, for example, can lose some of their concentration before even reaching the bloodstream, as they are broken down in the digestive tract. Once the drug enters the blood, it will be carried to all the organs in the body. Organs such as the liver and the kidneys will remove the drug from the body by metabolising (breaking down) and/or excreting it.
The term describing this loss of the drug is bioavailability. This provides information on the amount of a drug dose that is available to carry out its desired function.
For a drug to be able to work and travel to where it is needed, it must first pass through numerous membranes in the body. This is known as drug transport. The drug can either be transported around the body through the cells (transcellular) or via the fluid in between the cells (paracellular).
A drug will start to produce its desired effect once its concentration at the target site reaches a certain critical value. Once this happens the drug will start to interact with compounds at the target site (proteins or DNA) which will then produce a response. The time taken for this to happen will vary with the drug and the size of the dose given. In general, the higher the dose, the shorter the time until the response.
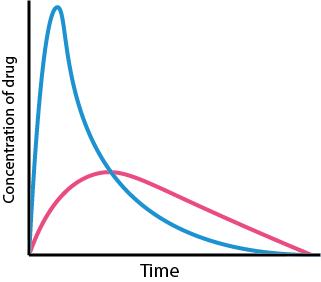
As can be seen from Figure 1, after taking a single dose the effect of the drug on the body will decrease over time. How long this will take depends on the drug and how quickly it is removed from the target site. Removal of the drug can happen by excretion, metabolism or redistribution to other parts of the body. The drug will continue to have an effect so long as its concentration at the target site exceeds a minimal effective concentration.
Drugs are designed so that they can provide successful treatment whilst giving minimal adverse side effects. Taking too little of the drug will mean that it won’t be able to produce its desired effect. Taking too much of the drug means that there will be too high a risk of adverse effects and so the risk of toxicity from the drug will outweigh the benefits. Every drug is toxic in high enough concentrations.
By controlling dose concentration and timing, drug concentrations can be kept inside a window that will produce successful therapy. The desired effect will be produced without causing too many adverse effects. This concentration range is called the therapeutic window and will be different for every drug (see Figure 2) and may vary from person to person.

Figure 2 shows that both the probability of the desired response (red trace) and the adverse response (blue trace) will increase with increasing drug concentrations. For each drug a decision will have been made as to what extent the beneficial effects of the drug outweigh the adverse ones. The answer will be very dependent on both the drug and the condition being treated. Obviously, drugs which are characterised by a wide therapeutic window are safer to use than those with a narrow therapeutic window (a well-known drug with a narrow therapeutic window is paracetamol).
Drugs will be prescribed to be taken as a fixed dose with a fixed time interval between doses. This means that the concentration of the drug in the body will be constantly changing, but will eventually rise towards a fixed value - known as a plateau. This is because each time a new dose is taken, only some of the drug from the previous dose will have left the body. This is illustrated in Figure 3.

Taking a dose of a drug too frequently will mean that it is more likely for the concentration to exceed the plateau (which has been designed as a safe level of a drug). This means that the level of the drug in the body may no longer be within the therapeutic window (see Figure 2) and so adverse effects may start to occur.
Drug delivery vehicles
The most common form of drug delivery is oral administration. It is favoured for its ease of use. However, it is one of the slowest and least efficient forms of drug delivery and many drugs cannot be administered this way. This is because drugs given orally must first pass through the digestive system before being absorbed. Some drugs are broken down during this process and are called “orally inactive”, and hence are administered in other ways.
Taking drugs orally can cause harm to the digestive system. Although tablets provide a consistent dosing, this does not account for the contents of the stomach. Taking drugs orally on a full stomach will reduce absorption of the drug, but taking them on an empty stomach can increase the risk of causing damage to the stomach lining and in some cases even lead to stomach ulcers (non-steroidal anti-inflammatory analgesics, such as ibuprofen or aspirin, can cause such problems). Some drugs (antibiotics) when taken orally can harm the beneficial bacteria in the gut, leading to a range of health implications.
When oral administration is not suitable or not desirable, suppositories can be used. The drug is administered in solid (soft, waxy) form into the rectum, urethra or vagina, where it melts and dissolves. Suppositories can be used when prolonged drug action is required. The major disadvantage of this method is that it is slightly inconvenient and can cause irritation at the site of administration. Absorption of drugs from suppositories can be unpredictable.
Another drug delivery method is transmucosal administration. The drug is used in the form of nasal sprays, gels or as quick dissolving tablets for oral use. The drug is administered either into the mouth or into the nose where it diffuses through the mucosa (tissue that lines the mouth or nose). Once diffused through the mucosa, the drug enters the bloodstream via the jugular veins (large veins in the neck, which carry blood from the head and face) from where it is delivered to its site of action. This method is suitable for patients who have difficulty swallowing, since the drug diffuses through the mucosa instead of having to be swallowed. This method also bypasses the digestive tract, avoiding the risks that were mentioned above for oral delivery. There is a small risk of accidentally swallowing the delivery system before it has had a chance to diffuse, which may render the treatment ineffective.
Drugs can also be administered via the nose or mouth in the form of an inhalant. Although drugs taken this way can spread to the rest of the body, this method is typically only used at the drug target (for example, decongestant nasal sprays). Inhalants are a very fast form of drug delivery but require a specialised delivery gadget (the inhaler itself) which must be able to administer the correct amount of drug each time.
In transcutaneous administration the drug is directly applied to the skin, for example as creams, ointments and skin patches. The active ingredients then dissolve through the skin and into the bloodstream. Advantages of this method are that prolonged and controlled drug release can be easily achieved, allowing for a constant level of the drug in the bloodstream, as well as being non-invasive. However, some people can have allergic reactions to the adhesive chemicals in skin patches which can cause contact dermatitis (irritation) at the site of application.
When drugs are injected they can be injected in one of three main ways (see Figure 4): intravenously (into a vein), intramuscularly (into a muscle) or subcutaneously (into a layer of fat and connective tissue under the skin). Intravenous delivery is the fastest method with drugs reaching their target in seconds, whereas intramuscular and subcutaneous injections provide a slower continuous release of a drug (with intramuscular delivery being faster than subcutaneous).
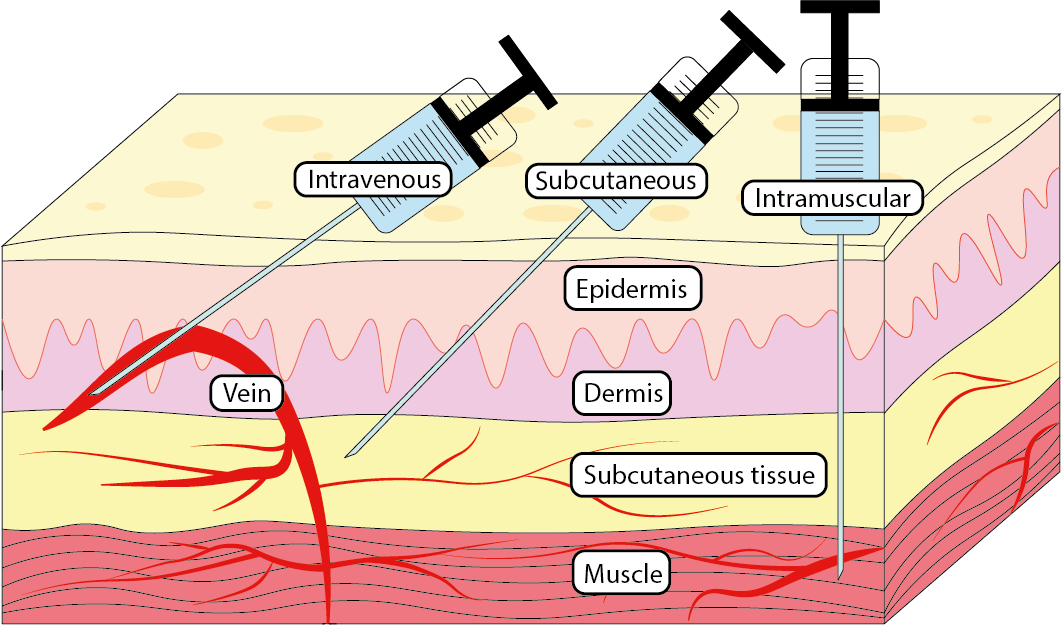
Injections are favoured for their efficiency and speed of delivery. Intravenous injections are essential in emergency situations when drugs may be needed immediately at a target site. Intramuscular and subcutaneous injections are very useful for a slower and more steady release of drugs and are suitable for drugs that are orally inactive such as insulin. Both intramuscular and subcutaneous injections can be self-administered in various ways and at different locations of the body.
Signals, either from the brain to the body (in the case of movement) or from the body to the brain (in the case of registering senses), are carried via long thin cells called neurons. Local anaesthetics temporarily disable the neurons’ ability to carry signals. This blockage is useful for preventing pain during operations avoiding general anaesthetics, or for treating other existing pain. Local anaesthetics can be administered as injections or topologically as a cream, gel or ointment.
Drug interactions
With most effects referred to as ‘drug interactions’ being unwanted or harmful effects, and with increasing numbers of people being on long-term medication with some times elaborate ‘drug cocktails’, interactions of drugs with other drugs, of drugs with food or drink as well as patient-specific responses are an increasingly important consideration for the safe management of medication plans.
Drug - drug interactions: this type of interaction occurs between different drugs, including prescribed and over the counter medications, herbal remedies and dietary supplements. Some examples of this type of interaction include:
· Warfarin and antibiotics – warfarin is a widely used anticoagulant (blood thinner) to reduce blood clotting; antibiotics overall are probably one of the most widely prescribed type of medications, for the treatment or prevention of bacterial infections. Some common antibiotics and antifungals (for example, metronidazole and fluconazole) enhance the blood-thinning effect of warfarin, leading to a significantly increased risk of major bleeding events or strokes. Antibiotics are not the only drugs that increase or lower the blood-thinning effect of warfarin, accordingly its effects need to be carefully monitored by regular blood tests (INR).
· Opioids and opioids and/or alcohol – a wide range of opioids are used as analgesics to treat moderate to severe pain, or are being used as recreational drugs. Examples are morphine or codeine, tramadol, cannabis (tretrahydrocannabinol, THC). These drugs are depressants, and so is alcohol, and these can mutually enhance these effects. Taking several opioids together and/or with alcohol can result in extreme drowsiness, impaired motor skills and lack of coordination. At higher doses this can lead to seizures, coma and death.
Drug – food / drink interactions: this type of interaction occurs between medications and food / drink. This should not be a big surprise, after all food and drink are metabolised in a variety of ways, many of which will at some stage lead to encounters with the metabolism of drugs, especially drugs taken orally. Some examples of this type of interaction include:
· Grapefruit - grapefruit (and some other citrus fruit) contains a type of chemical, furanocoumarins, that inhibits an enzyme (cytochrome P450) that is involved with metabolising (breaking down) many drugs in the liver and in the intestines. More than 50 drugs are known to be affected (when taken orally). Because of inhibiting this enzyme, the blood concentration levels of these drugs are increased, sometimes to dangerous levels, thus increasing the risk of serious adverse effects, depending on which drug is insufficiently metabolised.
· Cheese and MAOI’s – MAOI’s (monoamine oxidase inhibitors) are drugs commonly used to treat Parkinson’s disease and depression. MAOI’s inhibit an enzyme that keeps the concentration of a naturally occurring amino acid, tyramine, at the correct concentration by metabolising any surplus of tyramine. While inhibiting this enzyme helps to reduce depression, it also leads to increased concentrations of tyramine in the blood stream. Tyramine plays an essential role in regulating the blood pressure. If tyramine is not sufficiently metabolised it can quickly reach dangerously high concentrations. Tyramine is found in many foods such as soy sauce, chocolate and particularly in aged cheeses such as Stilton. Eating foods rich in tyramine while taking MAOI’s can lead to potentially fatal spikes in blood pressure. This effect is known as the ‘cheese reaction’.
Drug - patient-specific interactions: this type of interactions can arise from the interaction between medication and a pre-existing condition, such as a genetic disposition, or a combination of acute and/or chronic conditions occurring simultaneously. Some example of this type of interaction include:
· Penicillin – many people are intolerant of the antibiotic penicillin and its derivatives (chemically closely related, similar drugs (beta-lactams)). The degrees of intolerance vary and range from effects such as skin rashes (urticaria), itchy eyes and some swelling of face, lips and tongue all the way to an extreme allergic reaction, anaphylactic shock, which can be life threatening. Around 5 in 10.000 penicillin treatments result in anaphylactic shock. An allergic reaction to penicillin is an acquired consequence of a previous exposure to the drug (possibly even in just minute quantities in food) where the immune system misread the drug molecules as harmful substance, producing and releasing a specific antibody (which on the next exposure to the drug triggers the allergic reaction, a protective overreaction of the immune system to a perceived harmful substance).
Other classes of drugs show a similar spectrum of adverse reactions, all the way to anaphylaxis. For example, many people react to the non-steroidal anti-inflammatory (NSAID) drug diclofenac (used to treat moderate pain and swelling from inflammation) with severe skin rashes but there are also cases of systemic anaphylactic shock. It is sometimes difficult to distinguish adverse reactions and degrees of intolerance from serious allergies.
· Codeine – codeine belongs to the class of opioid analgesics and is used to treat mild to moderately severe pain. Codeine is a pro-drug and first needs to be converted in the body into its active form (morphine). Codeine fails to work as an analgesic in about 5-10 % of the population whereas another 5-10 % of the population metabolise considerably above-average amounts of codeine to morphine. The reason for these deviations from the ‘norm’ are differences in genetic make-up (genotypes). An enzyme in the liver, CYP2DC, is responsible for the codeine metabolism (amongst other substances) and to convert it to morphine. This enzyme is over- or under-expressed in a proportion of the population due to some genetic peculiarities.
· Placebo effect – an interesting, and in some regards, debated effect of the relationship between body and mind. The placebo effect summarizes the contribution(s) of somebody’s expectations on the outcome of taking some medication, or of some other interventions. If somebody expects adverse side effects from taking a drug, say nausea and vomiting, covertly giving this person a sugar tablet can trigger exactly these effects even if the tablet is completely devoid of active ingredients. If somebody expects pain relief from a drug, a covertly administered placebo can lead to pain relief. In some such cases, it has been shown that the placebo effect leads to physical changes. For example, expected and imagined pain relief increases the production of endorphins, the body’s own version of opioid drugs.