Other tumours
General aspects
All of the soft tissue sarcomas listed (see below) can occur anywhere in the body, including the head and neck region. Soft tissue sarcomas are rare but aggressive malignancies with an overall incidence of 5 per 100.000 per year and accounting for less than 1 % of all malignancies in the head and neck region. There are some notable exceptions and special considerations regarding this heterogeneous group (there are more than 50 histological subtypes known) of uncommon soft tissue malignancies as far as the maxillofacial region is concerned.
Soft tissue sarcomas display a considerable variation in their histology and clinical behaviour, similar to the situation observed for bone sarcomas (also rare as primary malignancies). The aetiology of soft tissue sarcomas is largely unknown but previous exposure to high-energy radiation, radiotherapy, is a well-known long-term risk for the development of secondary malignancies later in life, including soft (and hard) tissue sarcomas. While it is reasonable to assume that most sarcoma cases arise sporadically, some rare forms of genetic predisposition (for example, Gardner’s syndrome or Li-Fraumeni syndrome), exposure to certain chemicals (urethane, polycyclic hydrocarbons, some polyethylene derivatives), immunosuppression and some virus infections (for example, HIV infection) are all thought to be associated with increased risk for soft tissue sarcomas. It is not uncommon for soft tissue sarcomas to be diagnosed only after metastases and the effects of these, for example in lungs or bones, caused a visit to a clinician.
It should be mentioned that it is difficult to obtain useful (that is, large enough and comparable) data sets for meaningful statistical analysis for all rare diseases. Soft tissue sarcomas of the maxillofacial region are no exception to this fundamental rule. Even in the absence of extensive statistical analyses (and general guidelines based on such data) it is not difficult to see that a main challenge in the treatment of maxillofacial soft tissue sarcomas often is to realise wide resection margins in the first-line surgical treatment of these lesions, given the anatomic and functional constraints in this region.
Liposarcoma
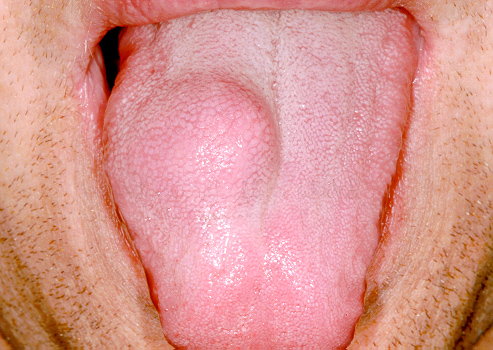
Only 4 % of liposarcomas are found in the head and neck region, yet liposarcoma is the most common soft tissue sarcoma in the maxillofacial region. The neck is the most common site, the lesion usually presents as a soft lump. The second most common site is the scalp. Liposarcomas in the head and neck region (see Figure 1) are thought to be significantly more often low-grade (less aggressive) malignancies than liposarcomas of other parts of the body. Simple excision and reconstruction has a reasonable high success rate.
Leiomyosarcoma
Leiomyosarcoma, originating from smooth muscle cells, is overall the most common soft tissue sarcoma. This type of smooth muscle tissue forms the walls of, for example, the intestines and the womb. Most commonly the stomach is affected by this type of sarcoma. Leiomyosarcoma is a rare tumour in the head and neck region but has been reported to occur in the nasal sinuses and the skull base. The low incidence of leiomyosarcoma in the head and neck region has been explained by the paucity of smooth muscle in this anatomic region.
Rhabdomyosarcoma
Rhabdomyosarcoma develops from striated muscle cells. It mostly affects children and adolescents, but is also seen in adults. Rhabdomyosarcoma is the most common soft tissue sarcoma in children and its most common location is in the maxillofacial region (near the eye, nasal sinuses, throat, palate and near the spine in the neck).
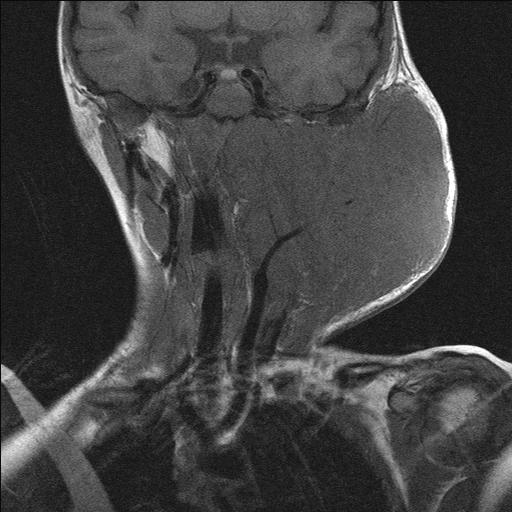
There are two main types of rhabdomyosarcoma, plus some rare variants. Embryonal rhabdomyosarcoma mainly affects young children but is the most common type at all ages. This variant is called ‘embryonal’ because of the characteristic appearance of the malignant cells, which resemble the developing muscle cells of a 6 to 8 week old embryo. The second main type, alveolar rhabdomyosarcoma occurs at all ages equally and accounts for approximately 20 to 30 % of rhabdomyosarcoma cases. It affects most commonly the head and neck region but also the extremities (see Figure 2). There does not seem to be a genetic predisposition for alveolar rhabdomyosarcoma.
Angiosarcoma
Angiosarcoma is the umbrella term for malignancies of the endothelium cell lines that form the wall of blood and lymphatic vessels (haemangiosarcoma refers to a malignancy of the blood vessels, lymphangiosarcoma refers to malignancies of the lymphatic vessels). Angiosarcomas tend to be aggressive malignancies with a propensity for metastasis.
Angiosarcomas affect men and women equally. They mostly affect the elderly and are very rarely seen in children. Most angiosarcoma cases belong to the category of cutaneous angiosarcoma (angiosarcoma of the skin), mostly in areas chronically exposed to UV light. Cutaneous angiosarcoma is usually found on the face or scalp.
Some angiosarcomas are found in deeper tissues (subcutaneous angiosarcoma). Cutaneous angiosarcomas usually show signs of bleeding and tissue necrosis.
Kaposi sarcoma
Kaposi sarcoma is a rare vascular malignancy that develops from the endothelial cells, similar to other forms of angiosarcoma (see above). In most cases it affects the blood vessels of the skin, mostly the upper and lower extremities and face, but also the mucosa of the head and neck region (most commonly the oral cavity, and there the palate, but has also been reported in the pharynx, larynx and tonsils).
Kaposi sarcoma can be caused by the human herpes virus 8 (HHV8) and in particular affects HIV infected people (nearly all HIV-positive people are also infected by HHV8) with high virus loads (such as AIDS; nowadays less common in the industrialised parts of the world). HHV8 infection is also strongly suspected to be related to the occurrence of some forms of lymphoma and Castleman’s disease (a systemic condition affecting the lymph nodes). Kaposi sarcoma is also found in people who are HHV8- and HIV-negative. Some types of Kaposi sarcoma seem to have some genetic predispositions, and most infected with HHV8 (common viral infection) never develop Kaposi sarcoma.
Kaposi sarcoma of the skin commonly starts with small, flat, slow-growing discoloured skin lesions. Kaposi lesions can also affect lymph nodes and other body organs (lungs and liver in particular). If the lymph nodes are affected, Kaposi sarcoma may cause painful lymphoedema (swellings caused by fluid retention).
Neurofibrosarcoma and malignant Schwannoma
The peripheral nerves are covered by a particular type of protective tissue that is made up of a cell type closely related to fatty tissue cells (Schwann cells (named after the physiologist T. Schwann), also called neurolemmocytes). This protective and insulating myelin sheath wrapped around the peripheral nerve fibres has multiple roles. It improves electrical conduction in neural signal transfer and aids general maintenance of healthy nerve tissue, and communication between body / muscle cells and neural impulses.
All peripheral nerves in the body can develop tumours of the protective sheath tissue, including the cranial nerves (in the skull), sympathetic chain (part of the autonomous nerve system, not under voluntary control; along the neck), brachial (nerve system running from lower front of neck to chest) or cervical plexus (serving the sensation and motion of the side and back of neck, head and scalp) in the head and neck region. One of the cranial nerves, the vagus nerve, is most commonly affected.
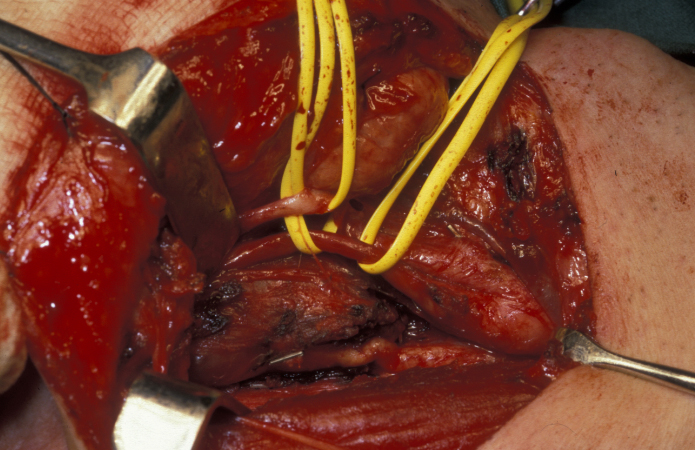
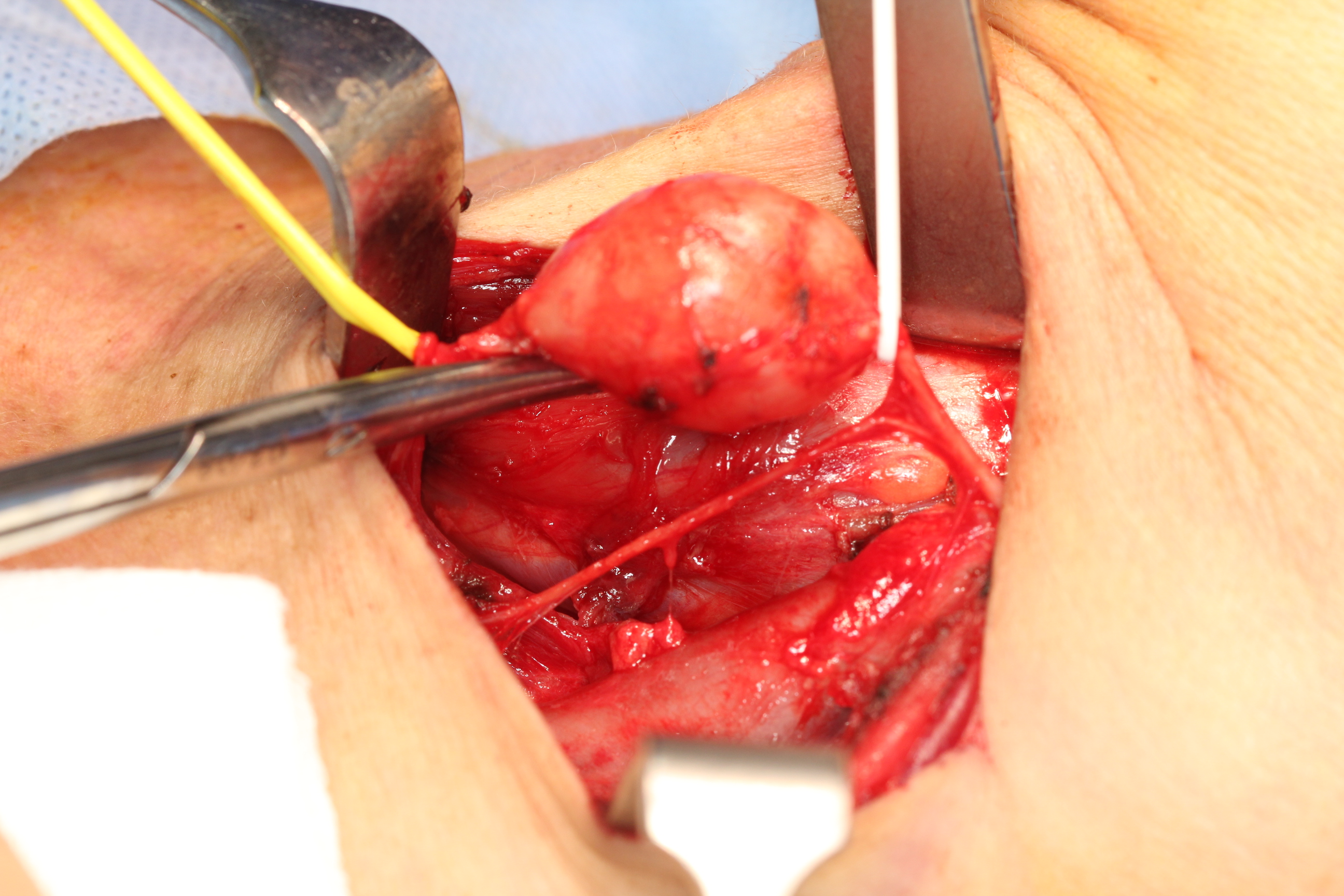
The majority of these neurogenic tumours (neurofibroma, Schwannoma) are slow-growing benign tumours. Approximately 25 to 40 % of these are found in the head and neck region. If possible / on balance, these tumours should be surgically removed: there is a small but significant risk of benign Schwannomas for malignant transformation (see Figure 3 and Figure 4).
Malignant peripheral nerve sheath tumours, neurofibrosarcoma and malignant Schwannomas, are rare aggressive malignancies with potential to form systemic metastases via haematogenous (blood flow) spread. Given the rarity of these tumours (fewer than 100 cases diagnosed each year in the UK), their aetiology is poorly understood.