Craniofacial anomalies
Contents
The diverse group of craniofacial anomalies comprises some of the most common birth defects in humans. Amongst these, a variety of jaw disproportion and cleft lip and/or palate conditions are the most common. These conditions are discussed in two separate sections:
This leaves us with a discussion of malformations affecting the skull and brain. All of these congenital craniofacial anomalies can be spontaneous developmental disorders or part of an inherited genetic syndrome (see below).
Craniosynostosis
The function of the skull (the cranium) is to protect the delicate brain tissue. This protective role requires finely matched size and shape of the hard shell and its soft content. This requirement in turn necessitates finely tuned sequences of development and growth of brain and skull bones, before and after birth.
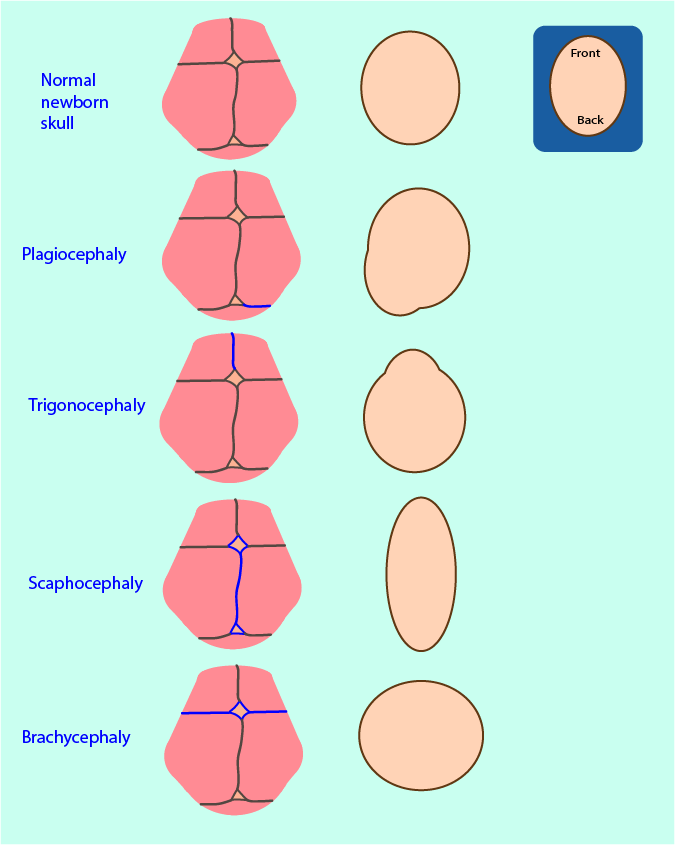
Nature has engineered a clever solution to accommodate growth of the brain while providing protection for it with a hard shell. Early in life, the skull is made up by bone compartments (the frontal, parietal, temporal, occipital and sphenoid bones) that are fairly flexibly connected by soft interfaces (sutures; see top row in Figure 1). Around the time of birth, the human skull is relatively small in order to enable easy passage through the birth canal. After birth there is an onset of rapid growth of the brain. The sutures of the skull permit corresponding skull growth perpendicular to their main direction. The sutures are geometrically perfectly arranged to allow for overall harmonious brain and facial growth.
Ongoing brain growth is the main factor that keeps the sutures open. Lack of soft-tissue growth signals biochemically that the soft sutures should fuse and harden, then turn into regular bone (ossification). This effect is desired once the brain has stopped growing in its normal developmental processes. It ensures mutually perfectly matched size and shape of brain and skull. In practice, fusion of the sutures is a gradual process correlated with the rate of growth of the brain and is complete by the age of 6 to 8 years.
Craniosynostosis is the result of one or several of the sutures fusing prematurely. It is called a primary craniosynostosis if it is not related to any other problems. It is called a secondary craniosynostosis if it is associated with some underlying condition. Secondary craniosynostoses are the more common. Microcephaly (an unusually small head at birth) is usually a strong indication for the presence of secondary craniosynostosis. Craniosynostosis may be simple (only one suture involved) or compound (multiple sutures involved). When only one suture is involved, usually the functions of brain and cranial nerves are not affected.
Early fusion can affect the sutures of the bone of the skull base or the vault. In either case, the growth space for the brain is hindered and compensatory growth (where possible) and distorted cranial and facial growth is the result. Some examples of deformities resulting from craniosynostosis of a single suture are schematically illustrated in Figure 1 by showing the effect as seen from a top view:
- (posterior) plagiocephaly as shown results from early unilateral fusion of the lambdoid suture;
- trigonocephaly results from early fusion of the metopic suture;
- scaphocephaly results from early fusion of the sagittal suture (the most common synostosis);
- brachycephaly as shown results from early bilateral fusion of the coronal sutures.
Multiple suture involvement tends to give rise to brachycephaly. The typical compensatory growth following fusion of the sagittal suture leads to an elongated shape of the head and bulging of the skull at the front and back.
Effects of craniosynostosis depend on the site(s) and extent of premature fusion; in more severe cases immediate and/or early paediatric craniofacial surgery may be required. The anomalies can have significant effects on cranial and facial growth (see Figure 2). Given the developmental course of events, craniofacial anomalies are usually diagnosed at birth or shortly afterwards.
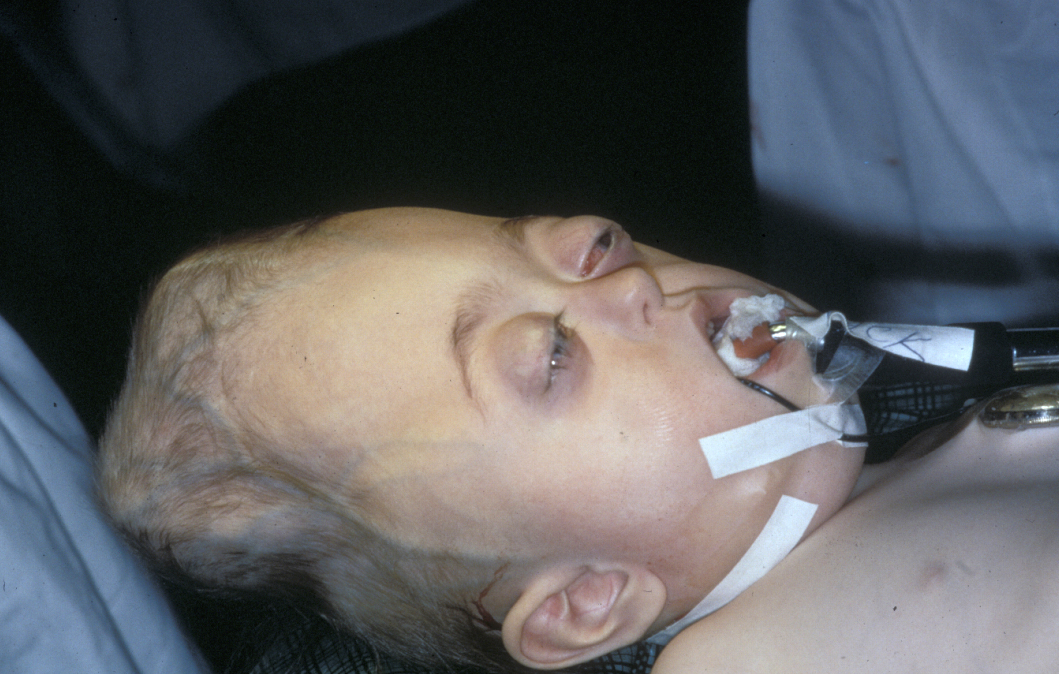
Restriction of growth of the skull vault can lead to a rise in intracranial pressure. This requires early intervention to decompensate and release pressure in order to prevent damage, for example to cranial nerves and potentially leading to loss of vision. Another neurological problem associated with craniosynostosis is a possible build-up of cerebrospinal fluid (hydrocephalus) that may necessitate the implementation of a shunt to reduce pressure and channel off surplus fluid. Craniosynostosis may also be associated with the displacement of the lower part of the brain (Chiari I malformation) toward the upper region of the neck spinal canal, causing build-up of pressure and ensuing damage in that region; urgent surgical intervention (removal of bone) to reduce pressure may be necessary.
Complex craniosynostoses typically occur as part of various craniofacial syndromes (see below). These conditions usually require early initial interventions such as securing the airway, combined with subsequent surgeries later in life.
Craniofacial microsomia
This is the second most common craniofacial anomaly, with a prevalence of approximately 1 in 5000 births. It is a congenital defect characterised by lack of both hard and soft tissues on the affected side of the face. It typically affects the mandibular ramus (the upward pointing branch of the lower jaw) and external ear (microtia). The majority of cases are unilateral, approximately 20 % of craniofacial microsomias are bilateral (affecting both sides of the face). In most cases craniofacial microsomia is associated with further malformations from underlying craniofacial syndromes (see below). Because of these common associations and a very wide range of suspected and reported chromosomal aberrations and other abnormalities, to date no specific genetic mutation has been identified as causal for microtia and microsomia.
A wide spectrum of ear and cranial nerve deformities is found. There may be mild abnormalities of the ear, or complete absence of the outer ear. In most unilateral cases of microtia hearing is normal on the non-affected side. Facial asymmetries may be mild and may be satisfactorily managed later with orthodontic treatment (such as hybrid functional appliances) alone. At the other end of the spectrum there may be significant defects such as complete absence or destruction of the mandible and/or temporomandibular joint (the joint connecting the lower jaw and the temporal bone). Such significant defects will typically require surgical intervention(s) often initially to secure the airway, later in order to restore functionality with regard to speech and oral food intake. Optimal timing of such reconstructive surgery is essential for long-term success.
Positional malformations
It is common for newborns to have a slightly strange head shape right after birth. Such minor distortions can arise from lack of space in the uterus or from the birth process. This all takes care of itself after a short while, usually within a year.
Sometimes a distortion of the head shape develops over time, with an appearance similar to plagiocephaly or brachycephaly (see Figure 1 above) but in the absence of any craniosynostosis. This is known as ‘positional moulding’ or ‘deformational plagiocephaly/brachycephaly’. It occurs if an infant spends much time either constantly lying on one side (plagiocephaly-like appearance), or on their back with the head on a hard surface (brachycephaly-like appearance). This kind of distortion is usually self-correcting over time, approximately two years, but recovery is helped by varying the positioning of the head of an infant.
Craniofacial syndromes
Craniofacial syndromes are genetic syndromes that can be isolated (spontaneous mutations) or inherited conditions. The associated abnormalities include a very wide variety of conditions and disorders, with approximately 700 such syndromes known (some of which are very rare). The disorders are not limited to conditions solely affecting the head (including craniosynostosis, cleft lip and/or palate, craniofacial microsomia, neuropathies of the cranial nerves, deficits of brain function) but may include further malformations of other parts of the body, often affecting the limbs, hands and fingers, as well as malformations of the heart and digestive system.
The embryonal development of head and face is a truly complicated course of events, involving multiple finely tuned cascades of biochemically / genetically encoded signalling, and so on. This delicacy of an amazing developmental process exposes the maxillofacial region to the effects of minor genetic mishaps and developmental disturbances (another example of such vulnerability to minor developmental disturbances is the common occurrence of cysts in the maxillofacial region). In the past, statistical analysis of family traits and similar investigations had only limited success in elucidating the pathogenesis and underlying mechanisms for craniofacial syndromes. The advent of genetic screening (see below) and other advances in molecular biology a couple of decades ago are now making a big difference in learning more about such conditions and their causes. Over the past few years several single and combined genes and their involvement in developmental disorders have been identified.
The most common (perhaps better termed: the least rare) craniofacial syndromes are summarised below; the severity of signs and symptoms varies widely among individuals.
Apert syndrome (autosomal dominant inheritance) occurs approximately in 1 : 75.000 births. The syndrome involves multiple prematurely fused sutures (coronal, sagittal, lambdoid), facial deformities including hypertelorism (wide eyes) and syndactyly (malformations of hands and feet). The syndrome is associated with a single gene mutation (FGFR2; involved with the physiology of bone formation).
Carpenter syndrome (autosomal recessive inheritance) is rare, with less than 100 cases reported in the literature. The syndrome involves multiple fused sutures with severe skull and facial deformities, malformations of hands and feet, cardiovascular deformities, skeletal abnormalities (such as scoliosis or deformed knee joints), early childhood onset of obesity, vision problems, and may include mental retardation. Mutations of two genes (RAB23 and MEGF8) have been associated with the syndrome; both genes are involved in various protein syntheses.
Crouzon syndrome (autosomal dominant inheritance) is reported to occur approximately in 1 : 63.000 births. The syndrome involves the premature fusion of the coronal and/or sagittal sutures. It leads to deformities of the midface, and vision and hearing problems. The syndrome may also lead to cleft lip and/or palate in conjunction with an underdeveloped maxilla (upper jaw) and abnormal dentition. The syndrome is associated with a single gene mutation (FGFR2).
Muenke syndrome (autosomal dominant inheritance) is reported to occur approximately in 1 : 30.000 births. The syndrome involves the premature fusion of the coronal suture. It leads to deformities of the midface and skull and is occasionally associated with an abnormally large head (macrocephaly). There may be hearing loss and delayed development or learning difficulties. The syndrome is associated with a single gene mutation (FGFR3; involved with the physiology of bone and brain tissue formation); some people who carry this gene mutation have no signs and symptoms of the syndrome.
Nager syndrome (unclear genetic inheritance; seems to occur more commonly as a spontaneous mutation) is a rare condition, only few cases (less than 100) have been reported in the literature. The syndrome involves micrognathia (underdeveloped lower jaw), malar hypoplasia (underdeveloped cheek bones) and cleft palate, leading to feeding and (potentially serious) breathing problems resulting from a blocked airway (due to micrognathia). There may be further deformities, mainly affecting the hands and fingers and the skeleton of the limbs. The syndrome seems to be associated (in some cases) with a gene mutation (SF3B4; mutation leading to faulty proteins involved in the formation of bone tissue) but there may be multifactorial causes, including environmental effects and mutations of further genes.
Pfeiffer syndrome (autosomal dominant inheritance) occurs approximately in 1 : 100.000 births. The syndrome involves multiple prematurely fused sutures. It leads to skull and facial deformities, hypertelorism, hearing loss and dental abnormalities. In addition, the syndrome leads to brachydactyly (short fingers and toes) and sometimes syndactyly. There are different subtypes of the syndrome, with variable degrees of severity of signs and symptoms. The syndrome is commonly associated with a single gene mutation (FGFR2).
Pierre Robin sequence (autosomal dominant inheritance, but more commonly a spontaneous mutation) occurs approximately in 1 : 10.000 – 15.000 births. It can occur as an isolated syndrome, or in combination with other syndromes (such as Stickler syndrome, see below). The syndrome is called a sequence because of the sequence of developmental events leading to the specific deformities: micrognathia (underdeveloped lower jaw) causes glossoptosis (misplaced tongue, toward the back) which in turn leads to cleft palate. The deformities can cause difficulties with breathing and feeding early in life but growth of the mandible (lower jaw) in many children will catch up later and result in normal-sized features in the adult.The syndrome is commonly associated with a mutation of the SOX9 gene but further genetic causes may be involved as well as non-genetic factors. The SOX9 gene is involved in protein synthesis essential in the development of tissues and organs of an embryo.
Seathre-Chotzen (usually autosomal dominant inheritance, but also spontaneous mutation) is estimated to occur in 1 : 25.000 – 50.000 births and its severity varies widely. The syndrome involves the premature fusion of the coronal suture and other cranial malformations. It leads to skull and facial deformities, including eyes and eyelids, as well as malformations of hands, fingers and toes. Mutation of the TWIST1 gene are associated with the syndrome. A faulty TWIST1 gene prevents the synthesis of a crucial protein for the embryonal development of bones and muscles in the head and neck region and the limbs. Not all carriers of the mutated TWIST1 gene show features of Seathre-Chotzen syndrome.
Stickler syndrome (some subtypes of the syndrome follow an autosomal dominant, some an autosomal recessive inheritance) is estimated to occur in approximately 1 : 10.000 births. It includes a group of syndromes that in turn all include the features of the Pierre Robin sequence (micrognathia, glossoptosis, cleft palate, see above). In addition, Stickler syndrome includes problems affecting the eyes such as glaucoma, cataracts, or retinal detachment, alongside extreme forms of myopia (short-sightedness) and additional problems with hearing loss. The syndrome also involves a range of malformations and other abnormalities of the skeleton, in particular the spine and joints (including scoliosis, kyphosis, hypermobile joints and early onset arthritis). Most subtypes of Stickler syndrome are associated with a mutation of the COL2A1 gene; defects of this gene prevent the formation of regular connective tissues (such as collagen).
Treacher Collins syndrome (autosomal dominant inheritance, but more commonly a spontaneous mutation; there is also an autosomal recessive inheritance variety) is estimated to occur in approximately 1 : 50.000 births and the severity of its signs and symptoms varies greatly. The syndrome mainly leads to midface deformities (including malar hypoplasia (underdeveloped cheek bones), micrognathia (underdeveloped lower jaw) and cleft palate, hypoplastic sinuses) such that often serious breathing problems are caused. There may also be hearing loss from ear deformities, but intellectual function is completely normal. Most cases of this syndrome are associated with a mutation of the TCOF1 gene; this gene is an important part of the machinery to produce ribosomal RNA (rRNA), which in turn is a crucial agent in protein synthesis and cell life-cycles in the body in general. Why a lack of rRNA resulting from this genetic mutation predominantly affects the facial development is not currently understood.
History, examination and diagnostic tests
Most craniosynostoses are identified before or at birth and worsen over time (whereas positional malformations also worsen over time but are absent at the time of birth; minor malformations acquired during the birth process do not worsen over time). The history of events before, during and after birth usually holds some clues about the nature and origin of the problem; history in this context also includes the relevant medical history of the family as there may be information about possible familial / inherited syndromes.
Examination includes all the usual physical examinations and measurements typically performed in the assessment of newborns and/or young babies and may be completed by a range of imaging methods (X-ray and CT scans provide information about prematurely fused sutures and other bone deformities, MRI scans are useful to find out about soft-tissue malformations, including Chiari malformation (see above) or other brain and nerve abnormalities). Depending on the overall condition, ophthalmologists and neurologists may be involved in the process.
Genetic screening plays in increasingly important and increasingly practical role in investigations of craniofacial malformations. The screening process usually includes three generations of a family, so can be quite extensive. It may be searching for a single particular mutation as well as, more commonly, searching more widely for a range of genes and their potential abnormalities. Genetic screening provides important information about the origin of a condition as an inherited syndrome or as resulting from a spontaneous mutation. Genetic information thus is important for families in order to understand the risk of recurrence for future children, but also to be aware of additional health issues and enhanced risks that may be associated with a particular existing syndrome.