Infection
Below we give short descriptions of the main mechanisms of action of the three categories of antibiotic agents: antibacterials, antifungals and antivirals.
Antibacterial agents
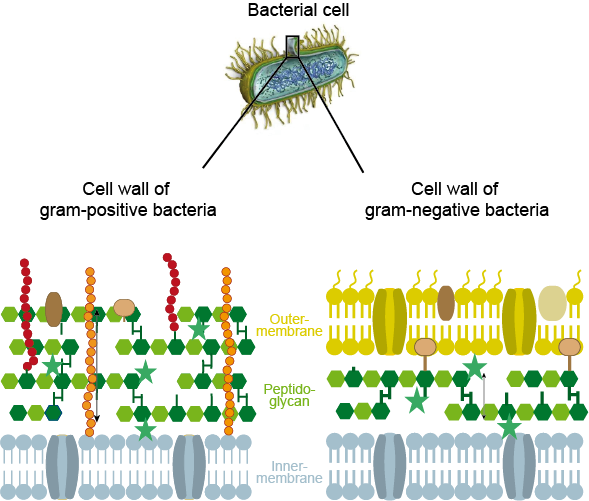
It can be advantageous to identify the type of bacteria responsible for an infection before starting treatment, given the large number of potentially harmful bacteria. For example, it can be useful to know whether bacteria belong to the Gram-positive or Gram-negative category and thus, because of their different types of cell walls (see Figure 1), will be susceptible to treatment with different types of antibacterial agents. It can also be useful to know if an infection is caused by anaerobic of aerobic bacteria.
Where it is not possible to precisely identify the relevant bacteria before starting treatment (which is the usual clinical scenario), it is common practice to start with a ‘best guess’, usually a so called broad-spectrum antibacterial drug (a drug that can treat a range of different bacterial infections). If later on additional information becomes available, the medication regimen can be adapted accordingly (in many maxillofacial infection cases, this is an ‘after the event’ scenario: by the time the pathogens have been identified, the infection has already been cleared).
The ways in which bacteria multiply, their structures and metabolic processes, through which they not only survive but thrive and replicate, are quite different from the metabolism and life cycles of the cells of mammals (including humans). These differences can be exploited for treatment with antibacterial drugs, which deny the bacterial cells some element(s) that are vital for their survival, whereas the host cells are only minimally compromised. The logic is to provide the host’s cells with a significant competitive advantage over the bacterial cells that have so far evaded the body’s immune system, which has resulted in infection.
There are essentially four main mechanisms by which antibacterial agents can interfere with bacterial cell function. The sketch of a bacterial cell in Figure 2 summarises these four mechanisms.
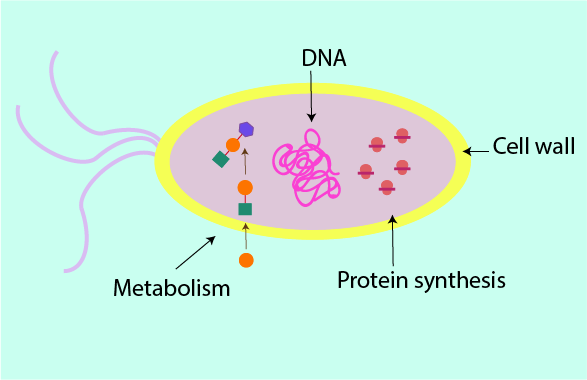
Some antibacterial agents kill bacterial cells outright (bactericidal agents), others weaken the bacterial cells and hinder their replication (bacteriostatic agents). The success of antibacterial treatments does not depend on this distinction. It is far more important to start treatment promptly, using the most suitable type of agent for the treatment of the particular pathogen(s) at hand.
Many mammals (including humans) and other living creatures naturally synthesise antibacterial substances in their normal metabolic processes to stay healthy (frogs, for example, continuously produce slime on their skin to keep their skin healthy; slime on frog skin contains several substances with antibacterial properties). Humans make lysozyme in saliva and other body fluids (tears); lysozyme is a highly effective antibacterial agent, it attacks bacterial cell walls (see below). Most / many of the existing synthetic man-made antibacterial agents are derived from naturally occurring substances and are probably better viewed as ‘discovered’ rather than ‘designed’ (although this may change in the future).
The bacterial cell wall
The cells of humans do not have a cell wall, only a membrane. In contrast, the strong cell wall of a bacterium is vital for its survival. The cell wall also makes a difference with regard to mobility of cells: bacterial cells, protected by their cell wall, are optimised for moving around and spreading in this way, human body cells are optimised to stay in place and carry out their various specialised tasks locally.
The bacterial cell wall needs to withstand large internal pressure changes within the bacterial cell, a consequence of the need to maintain a delicate osmotic balance (water flowing in and out of the cell). The cell wall also maintains the integrity of the cell membrane without which the controlled exchange of chemicals (homeostasis) between the bacterial cell and its surroundings is compromised. This vital protective role of the cell wall is enabled by a structural protein, peptidoglycan, which forms the main structural component of a bacterial cell wall. Bacteria have to synthesise peptidoglycan for their cell walls themselves.
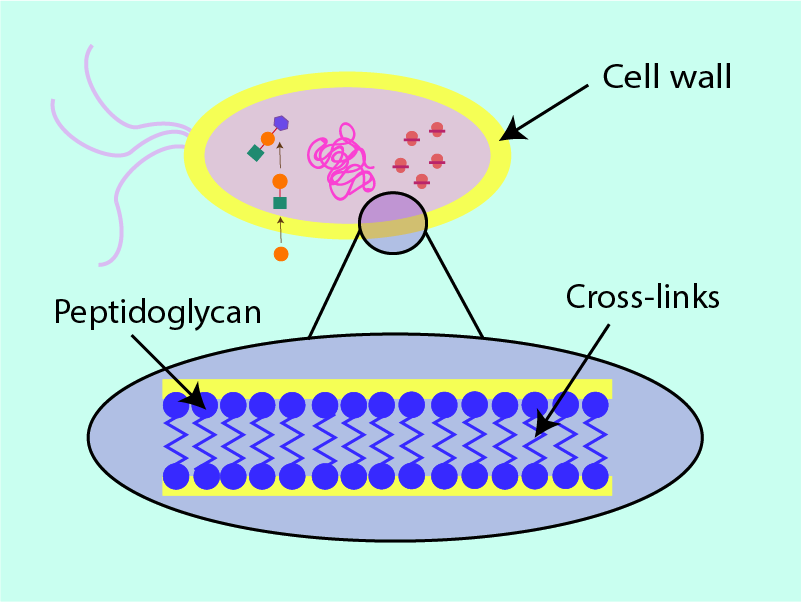
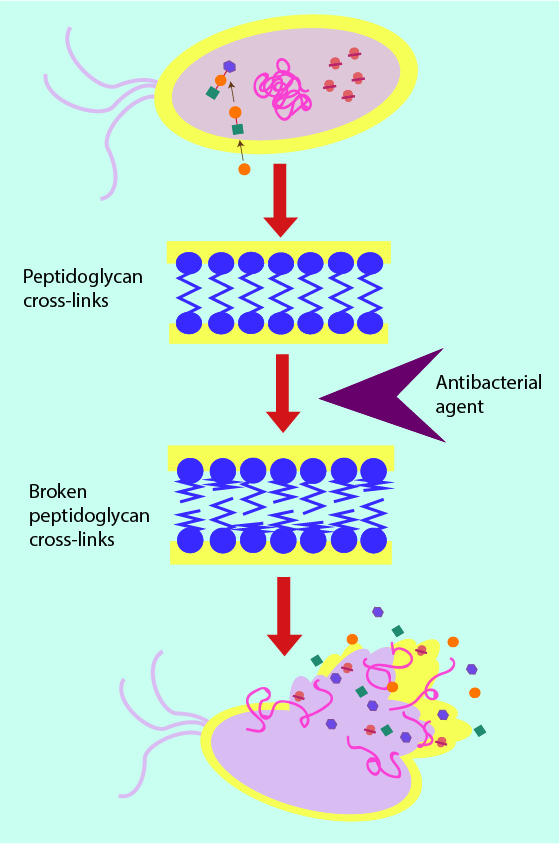
Figure 3 shows a cross section of peptidoglycan within the cell wall; long strands of the structural protein are interlinked with shorter strands. It is this ‘cross hatching’ (analogous to the supporting beams in fencing) which improves the ability of the cell wall to withstand pressure: the more crosslinks, the better the security and stability of the cell. Removal or weakening of these links by antibacterial agents weakens the cell wall by putting holes in the structure and thus leaving it susceptible to cell death. In a further analogy, the cell wall can be thought of as a dam of sorts. Antibacterial agents that target the cell wall can be thought of as weakening and putting holes in the structure of the dam, eventually causing water to flood through the wall into the cell and causing death of the bacterial cell. Most antibacterial agents acting on the cell-wall structures achieve this weakening effect by attaching themselves to the cell wall and creating weak spots, either for the agent itself to break the cell wall (for example lysozyme works in this way; see above) or to enable other chemicals (enzymes) to do so. This mechanism is illustrated in Figure 4.
Some metabolic processes of the bacterial cell
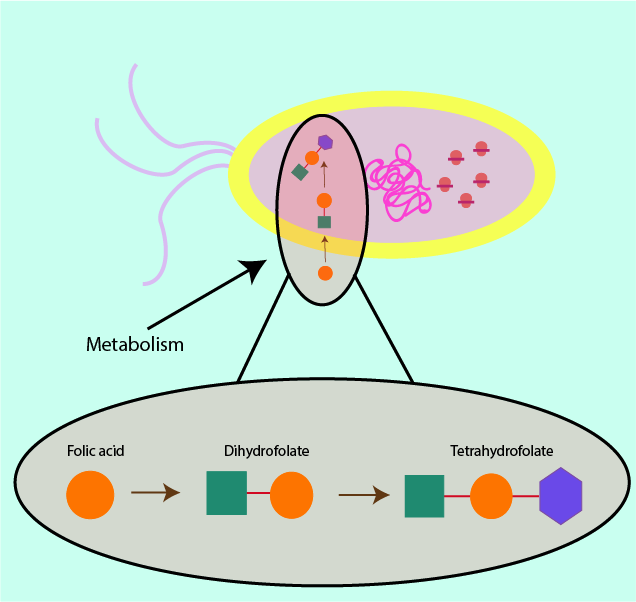
Some metabolically important substances are available to humans from dietary sources, whereas bacteria have to produce these substances for themselves via their metabolic processes. A sketch of a metabolic process is shown in Figure 5; smaller chemical units are put together to build bigger, more useful molecules for the cell. If a bacterial cell has to synthesise (make) a molecule in order to survive, then hampering this production process will have the same effect as starvation.
Where it is possible to deny a bacterial cell the necessary nutrients and resources, this will ultimately lead to the death of the bacterial cell by starvation. An example of a chemical which is taken in through the diet in humans but has to be synthesised by bacterial cells is tetrahydrofolate, a part of the vitamin B cycle (see Figure 5). Preventing tetrahydrofolate production via inhibition of folic acid production in the bacterial cell leads to a deficiency of that molecule in the bacterial cell and the ultimate purpose of the product remains unexecuted. Folic acid is important in the making of DNA (in all cells), so prevention of its synthesis in the bacterial cell makes cell death imminent as the cell can no longer replicate DNA.
In a slightly different mechanism which also interferes with the metabolic processes of the bacterial cells, some antibacterials can mimic the structure of molecules needed by the cell and resulting in their uptake instead of the normal molecule. This is schematically illustrated in Figures 6. It results in a deficiency as the cell fails to uptake and/or synthesise the correct and necessary molecule for its survival. This ‘pretender’ mechanism is also exploited in a range of drugs used in conventional chemotherapy, where for example, 5-fluorouracil is administered systemically and replaces uracil as the normal substrate in the cell metabolism.
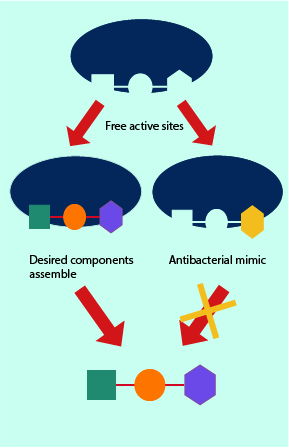
Once a bacterial cell has assembled a larger molecule, proper functionality requires this molecule to fit into the chemical and biological machinery of the cell. The molecule and the machinery often have complementary shapes, they match up well and any disruptions in this matching process results in the machinery being unable to function. ‘Pretender’ molecules smuggled into the cell’s metabolic pathways can either block the synthetic pathway from complete execution, or may lead to final products that are mismatched and thus cannot fulfil their specific biochemical task.
The protein synthesis in the bacterial cell
Synthesising proteins in all cells is much like following a recipe. Proteins are made from small molecular building blocks (amino acids). A bacterial cell has all the cellular equipment and machinery it needs as well as a recipe to follow, encoded in the form of DNA molecules. DNA is essentially the cell’s list of instructions as to how and when to make proteins, and is even able to go so far as to dictate how much of a protein is needed (depending on its function/importance within the cell). A part of the machinery necessary for protein synthesis in a cell is a structure known as a ribosome (a very large molecule), comprised of a small compartment and a large compartment.
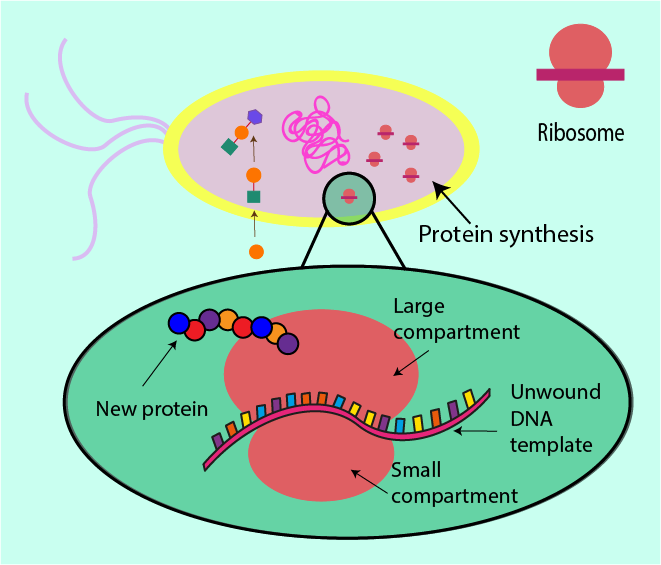
Before DNA can be ‘read’ by a ribosome, it needs first to be converted into a different language, which the ribosome can more easily and readily interpret. This translation is the function of another large molecular structure which goes by the name of RNA polymerase. RNA polymerase can simply be thought of as a middleman responsible for simplification of the recipe. Rewriting the recipe as a long linear line of instructions is easier to follow than the original message encoded in the twisted structure of DNA.
This more accessible message is then read by the ribosome which uses the string of instructions as a template to put together a protein: the ribosome clamps the instructions between its two compartments and moves along reading it as it moves, simultaneously producing the protein defined by the instruction. Figure 8 contrasts this ribosome action in normal circumstances with the situation where an antibacterial agent interferes with the role of the ribosome.
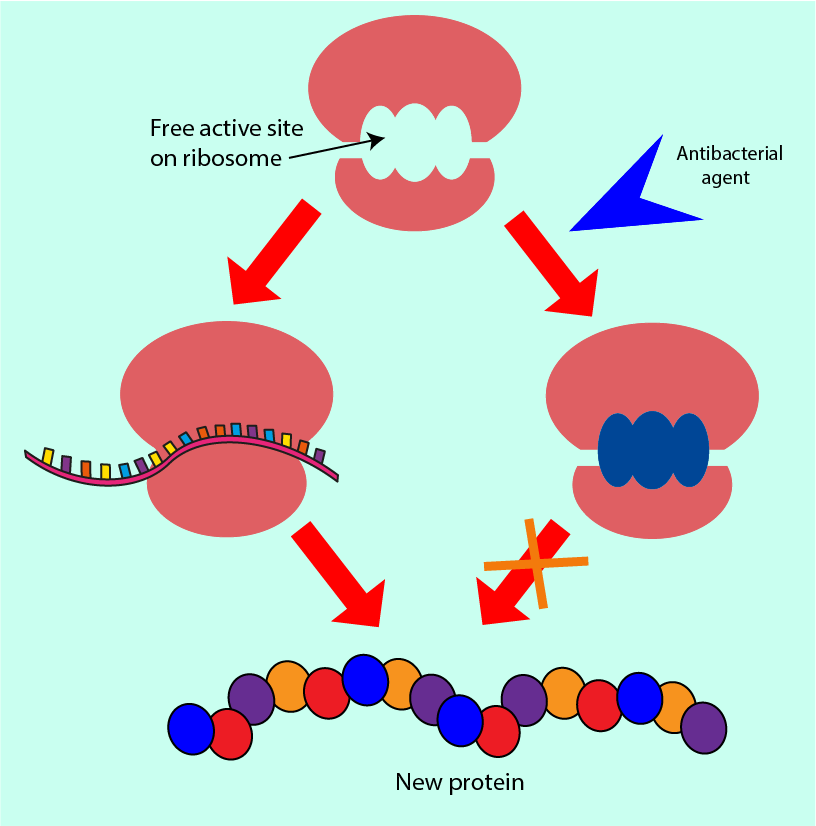
Antibacterial agents which interfere with protein synthesis can block the reading space within the ribosome in which the instructions would normally bind. Alternatively, they can prevent other molecules from binding to the ribosome (molecules that would normally help it to read the instructions more efficiently). Some antibacterial agents in this group allow the initial binding of the ribosome to the instructions but cause the machinery to misread the instructions, producing partially or completely dysfunctional proteins. All of these interferences mean that the bacterial cell ribosomes do not produce the correct functional proteins that the cell would need in order to survive.
Bacterial DNA
DNA is considered to be the recipe book for the cell’s protein syntheses. The DNA contains in an encrypted way (quite like a computer algorithm) the instructions for every protein the cell will ever need. However, the coiled and helical structure of the DNA keeps this message protected and stable within the cell so that it is not damaged as it is passed on during replication from cell to cell, from generation to generation. This is true for bacterial cells as well as for cells of mammals (including humans).
In order to synthesise new DNA for cell replication, the DNA has to be unwound from this protective supercoil arrangement in order for its instructions to be ‘read’, replicated and afterwards rewound. Essential worker molecules (enzymes) carry out these biochemical roles within a cell.
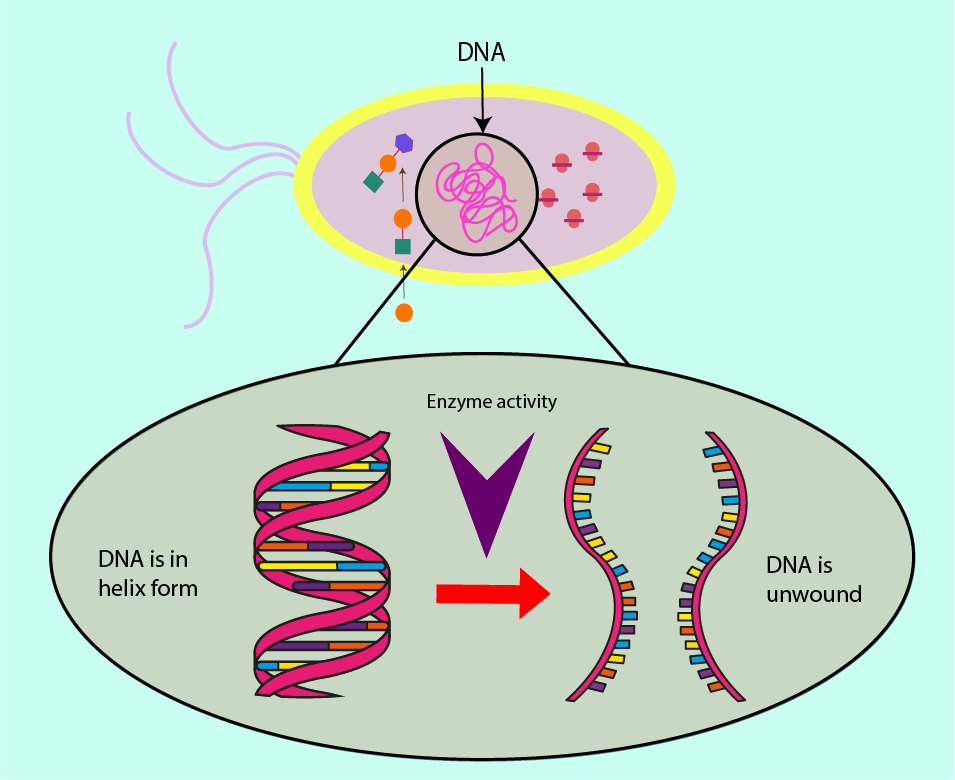
Two such enzymes, RNA polymerase and topoisomerase II, are targets for antibacterial agents.
RNA polymerase is responsible for using template strands of DNA to produce complementary strands of DNA, hence increasing the amount of DNA in the cell at every generation. Enzymes have a lock and key function, with the enzyme being the lock, and the key in this analogy is the molecule, or set of molecules, to which RNA polymerase binds. RNA polymerase is necessary for DNA replication. Preventing RNA polymerase from binding, either by altering the shape of RNA polymerase or introducing molecules to prevent the DNA from binding to it are mechanisms underlying antibacterial effects. As all bacterial (and host) cells contain DNA, interfering with the DNA synthesis can be considered as a ‘broad spectrum’ action of an antibacterial agent. Figure 10 symbolises this mechanism of interference with DNA synthesis.
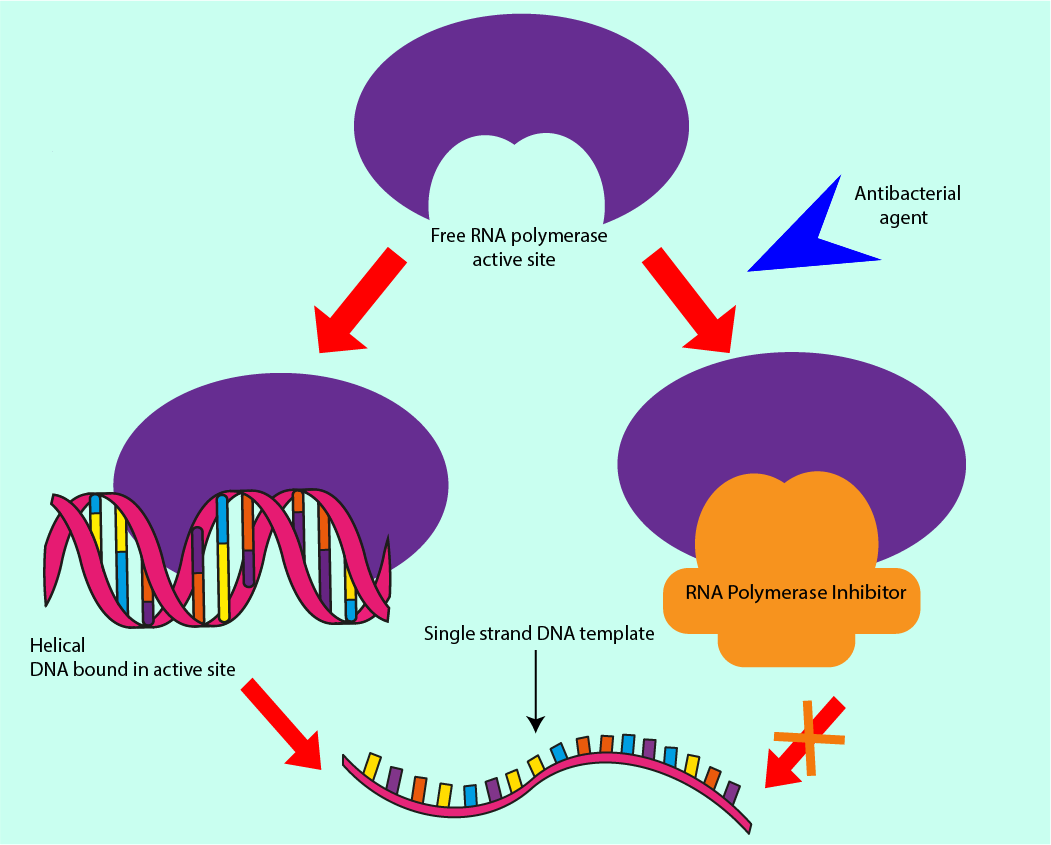
As well as interfering with the production of new DNA molecules, access to the template strands can be denied through inhibiting another enzyme, topoisomerase II. This enzyme is responsible for the supercoiling of DNA. Failure to regulate this winding of DNA can cause a build-up of torsion in the (fragile) DNA molecule, destabilising it and preventing the RNA polymerase from moving along the template strand, thus preventing synthesis of new DNA molecules. Not only the cell machinery responsible for making new DNA strands is vulnerable to interference(s) by antibacterial agents but also those molecules which regulate the process are susceptible to obstruction/hindrance (again, similar mechanisms of interference are exploited in some chemotherapy drugs).
Antifungal agents
Fungal infections have become more common over recent years. Factors contributing include an increase in the (over)use of broad-spectrum antibacterial agents, steroid inhalers, and larger numbers of people living with weakened immune systems (immuno-compromised) either after organ transplants, or during cancer treatments by chemo- or radiotherapy, or by living with HIV infection, or simply old age. A general reduction in commensal (useful) bacteria in the body not only makes infection by other, more harmful, bacteria easier, it makes it also easier for fungi to cause infection in an opportunistic way.
Similar to the philosophy of exploiting various different working mechanisms in antibacterial drugs, there are also four main mechanisms of action of antifungal agents. These four main mechanisms are summarised and symbolised in the sketch in Figure 11. There are further similarities between antibacterial and antifungal agents: several of these substances display both antibacterial and antifungal properties. This can be explained by some of the structural and functional similarities between bacteria and fungi.
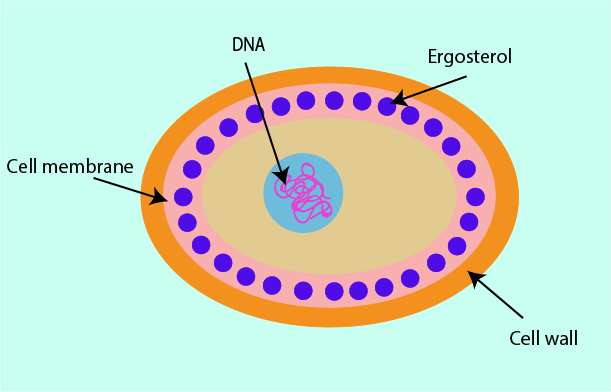
Cell membrane
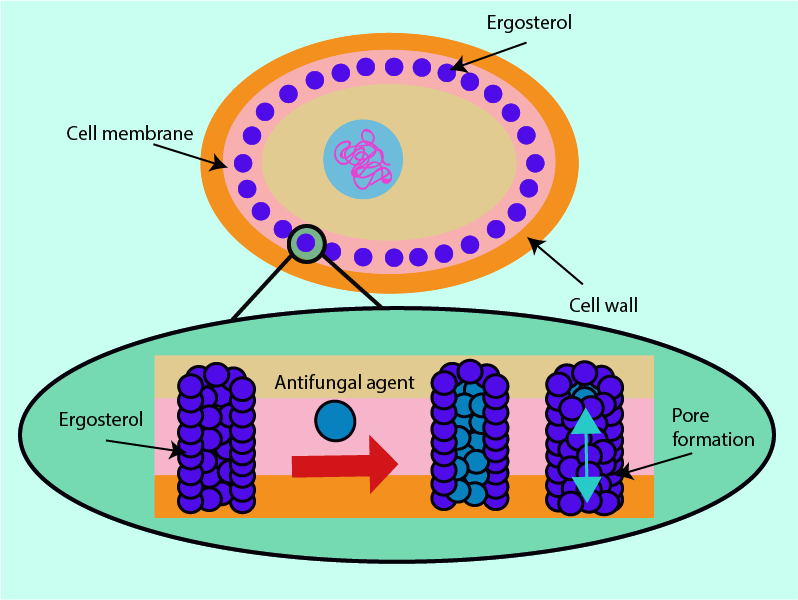
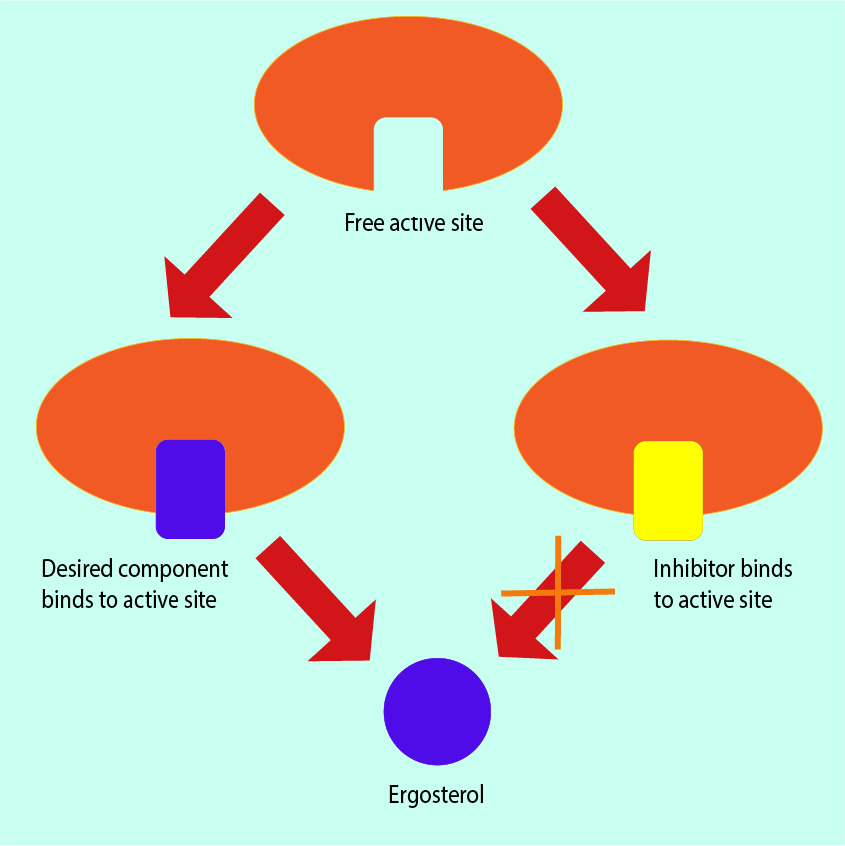
The cell membranes of fungi contain a structurally and functionally important molecule, ergosterol. It works in the same ways as cholesterol works in the cell membranes of humans but ergosterol is not present in human cells. This makes it a useful structure at which to direct antifungal drugs.
Some antifungals can bind with ergosterol. This results in the formation of holes or pores in the cell membrane structure; these holes have an effect similar to inserting straws into the membrane. These ‘straws’ (ion channels) give fluid (cytoplasm) normally contained within the cell an outlet from which the contents of the cell can leak out, causing cell death.
Fungal DNA
Some antifungals, upon entering the fungal cell, are converted into 5-fluorouracil, a chemical which competitively inhibits uracil from being included in newly synthesised DNA and RNA (uracil is one of the four basic DNA building blocks). Medicinal drugs that need to be converted into their chemically active form after delivery to their site of action of action are called pro-drugs (codeine is a well-known example – it needs to be converted to morphine in the body).
As an inhibitor of normal DNA and RNA synthesis, 5-fluorouracil causes cellular growth imbalances and ultimately the death of the fungal cells affected. In a different setting and different (systemic) drug delivery, the same underlying mechanism is exploited in the role of 5-fluorouracil as a chemotherapy drug.
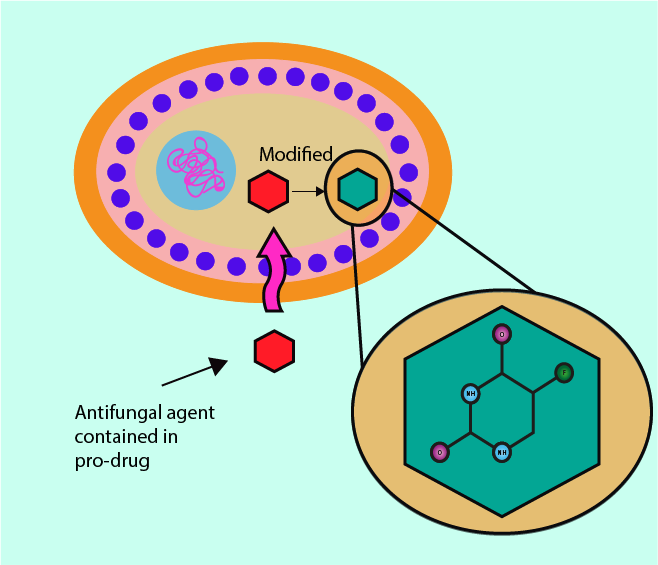
Metabolic inhibition
Targeting different stages of the ergosterol synthesis pathway (see above) always yields the same overall result: production of ergosterol is prevented. For example, some specific enzymes within fungal cells (p450 enzymes) are necessary to produce ergosterol. Many antifungal agents can bind with the p450 enzymes, thus blocking the ergosterol biosynthesis pathway. Because this inhibition mechanism is common amongst antifungals and p450 enzymes are common to fungi, antifungal agents that reduce the production of ergosterol are classed as broad-spectrum antifungals. Figure 14 illustrates this metabolic inhibition mechanism.
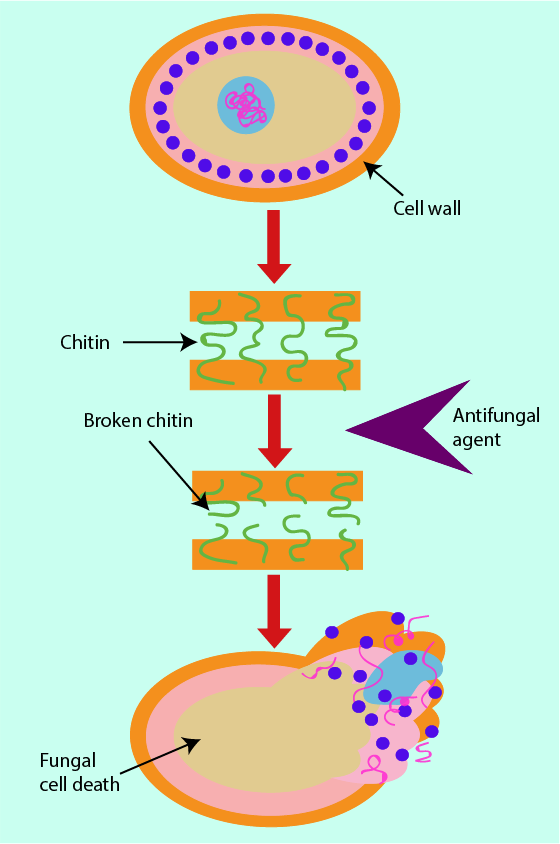
Cell Wall
The building blocks of bacterial cell walls are a target for the action of some antibacterial agents (see above). Similarly, there are structural building blocks unique to the cell walls of fungi that can serve as a target for antifungal agents. In fungal cell walls, the critical structural unit is a substance called chitin (a fibre-forming polysaccharide). Any antifungals that prevent chitin from forming in the cell wall cause a weakening of the fungal cell wall, eventually resulting in cell death (see Figure 15). Chitin is a chemical not found in human cells. It may thus be advantageous to use antifungal drugs that target chitin formation as this may minimise unwanted effects on the host’s system.
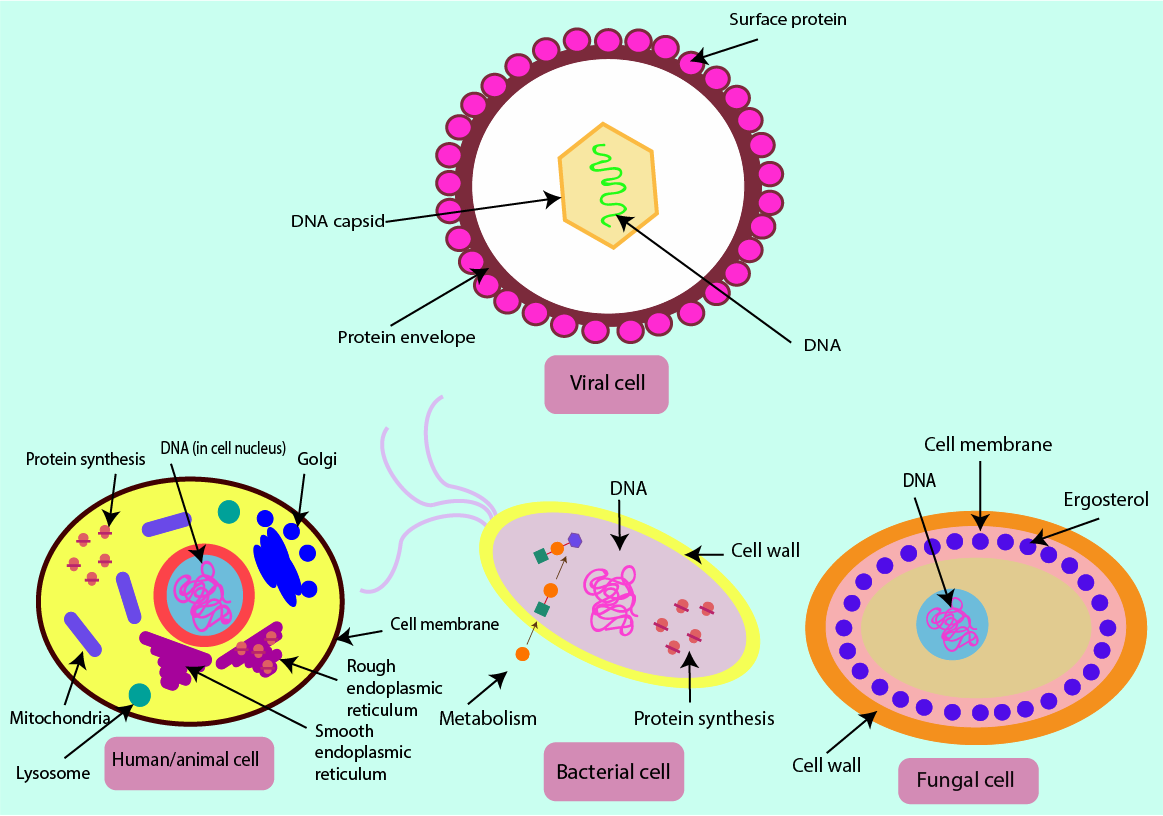
Antiviral agents
The structures and functionalities of viruses are even further from those of our own cells than bacterial or fungal cells. Viruses are made up of a single piece of DNA or RNA contained within a coating made of proteins (a capsid). Unlike bacterial and fungal cells, there are no cell walls, cell membranes or protein-synthesising machinery in viral cells. Accordingly, the working mechanisms of antiviral drugs will have to be different from those of antibacterial or antifungal agents. Figure 16 summarises the four types of cells and their main features. Figure 16 also serves as a reminder that there is no benefit from taking antibacterial drugs when suffering from a trivial viral infection, such as a common cold or sore throat (quite the opposite is true – it only comes with adverse effects of antibacterial agents on the host).
In order to reproduce (replicate) viruses have to take over the cells of the host. They need to ‘borrow’ protein-synthesis machinery and other cellular components from the host. They can do this either by getting inside the host cell, or by inserting just their viral DNA into the cell. The result of this invasion is that human host cells are (ab)used as factories for the production of new viruses. These newly produced viruses are released when the infected host cell dies. This close and direct dependence of the infectious agent, the virus, with the structure and function of the host cells explains why it is so difficult to develop drugs that only, or mainly, act on the virus and not the human cells (see Figure 17).
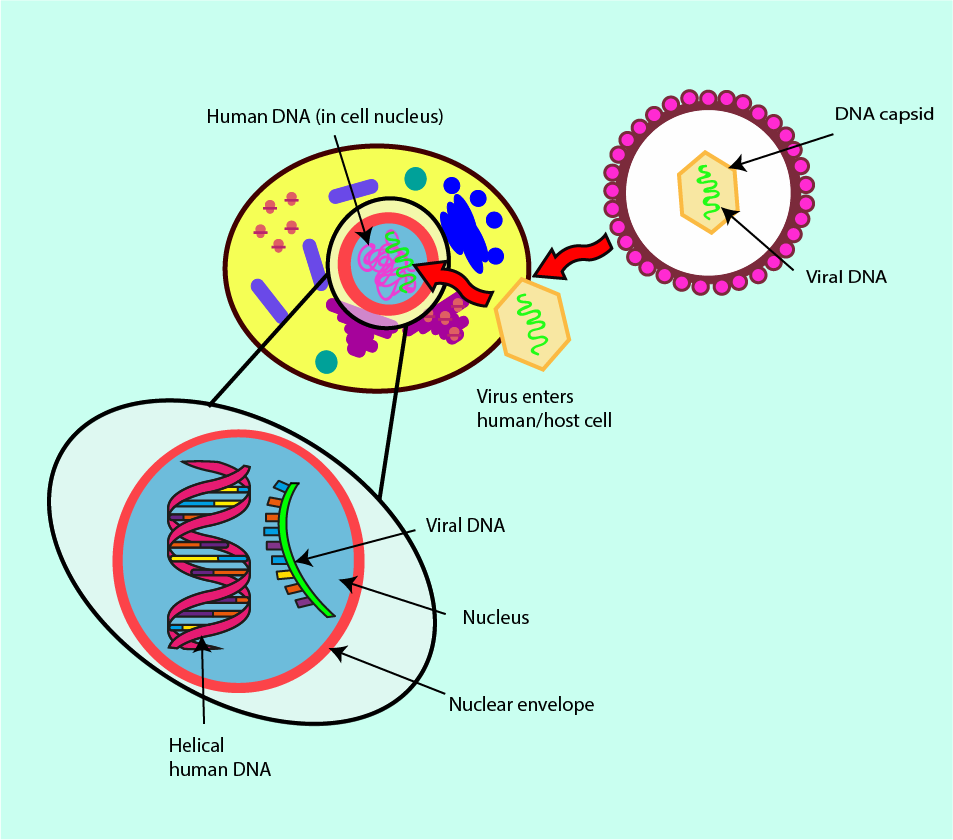
The ability of viruses to change (mutate) their protein coat further complicates the task of finding viable antiviral agents. The proteins making up the viral capsid can be used as drug targets. However, if the proteins, and thus the shape of the virus capsid, change rapidly any newly developed, or already established, antiviral drugs may miss the target. The molecular protein structures that once were a suitable and susceptible target for a particular drug may no longer be expressed by the virus strain and the originally designed drug will have lost its bite.
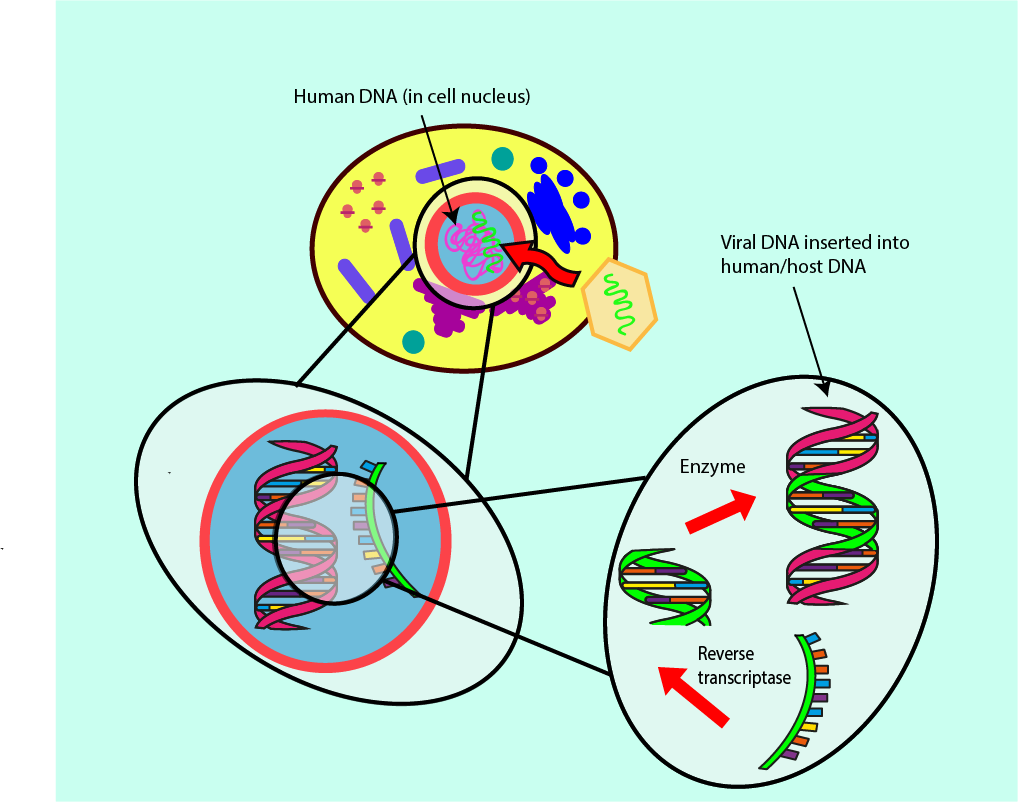
A subgroup of infectious viruses survives by following an even more integrated invasion path within the host cells. After infection, these viruses live within the DNA of the host cells and thus cannot be completely removed from there. The best-known example of this kind of retrovirus is probably the human immunodeficiency virus, HIV. Treatment options for retrovirus infections require careful consideration of antiviral agents which mainly influence the viral load and less so the DNA of the host. Figure 18 symbolises the infection mechanism and its integration in the host DNA by a retrovirus.
Below we outline the main working principles of antiviral drugs currently used to treat infections with a retrovirus and a ‘normal’ virus. Medication treatment for ‘normal’ viral infections is far less widely used than medications for bacterial or fungal infections.
Retrovirus antiviral agents
There are several enzymes, which participate in the task of integrating the viral DNA into the DNA in the host cell (for example, reverse transcriptase; see Figure 18). Preventing this function of reverse transcriptase means the viral DNA will not be integrated into the DNA of the host cell. Antivirals developed for treatment of retroviruses such as HIV focus on reverse transcriptase inhibition.
In order to convert the DNA of a virus into a form that fits the host’s DNA, the reverse transcriptase molecule requires some additional smaller building blocks, called deoxynucleotides, which occur naturally in the host cell. Some antiviral agents that are ‘pretender’ molecules (viral DNA base mimics) mimic the building blocks that reverse transcriptase usually bonds with, except for a few small but important differences. This mimicry means that the reverse transcriptase cannot build stable or lasting structures, resulting in a decrease in the rate of infection by the virus. Other inhibitors of reverse transcriptase interfere directly with the main part of its structure through which it binds to the viral DNA.
Protease inhibitors are another type of antiviral agents that reduce the virus load from infection with a retrovirus. After the viral DNA has been integrated into the DNA of the host cell, the next step of the infection cycle is the building of new viruses from this new instruction template imposed on the host DNA. The template is read by the host cells’ protein-making machinery (the ribosome; see above) to serve the needs of virus replication. The final resulting proteins need to be cut free from the template before they can serve their function. This is done by enzymes called proteases. Antivirals can prevent this final ‘cutting’ step by blocking the part of the cutter enzyme which attaches to the virus proteins. Functional versions of the virus proteins, made in the host cell, are then not processed and no new viruses will be assembled and released.
Other antiviral agents (targeting ‘normal’ viruses)
Some antiviral drugs, effective in treating infections with ‘normal’ viruses, work by stopping new viruses from escaping from the host cell. New viruses (virions) are not released from the host cell because an enzyme (neuroaminidase) is blocked by these agents. Neuroaminidase promotes the movement of virions, and blocking this enzyme thus reduces or stops the release of virions from the host cell.
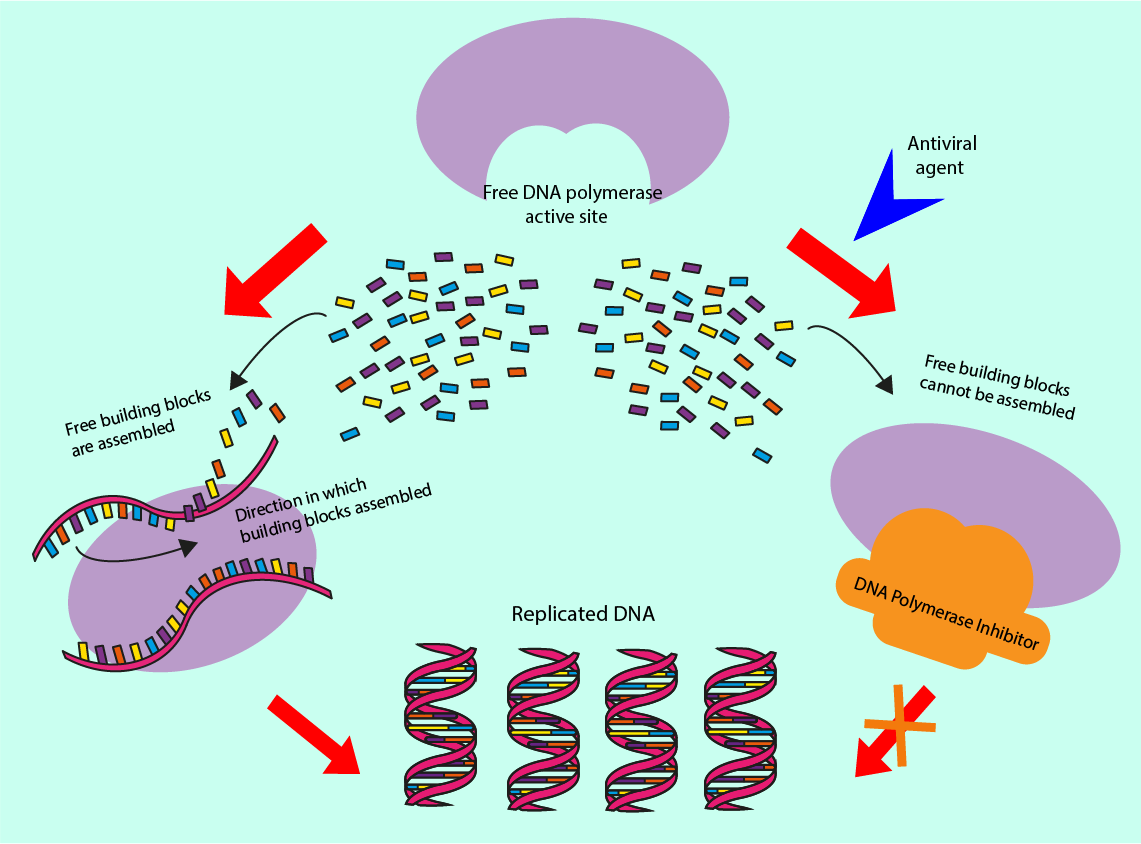
Other antiviral drugs prevent viral DNA replication and so prevent the virus from replicating. Antiviral drugs achieve this by inhibiting a virus-specific enzyme, DNA polymerase, by preventing it from binding to the viral DNA. Another such inhibition mechanism blocks other enzymes from binding to the DNA polymerase in the replication process. These two mechanisms are illustrated in Figure 19.
The human body commands powerful protective mechanisms of its own against viral infections, the immune system. Stimulating an immune response is an indirect mechanism by which some drugs boost the number and / or activity of the body’s own antiviral agents. This includes the boosting of enzymes that break down virus structures in the blood stream and immunoglobulins . A range of different immunoglobulins are synthesised by the immune system as a specific response to infections. These immunoglobulin molecules continue to circulate in the blood stream and specialise in recognising and inactivating specific virus cells (immunoglobulins are also responsible for long-term immunity against certain infections). Vaccines (see below) are a particularly relevant way to stimulate an often long-lasting immune response to viral infections.
Dealing with SARS-COV-2 and COVID-19
Given that at the time of writing (autumn 2020) there is no known cure for COVID-19 and no vaccine to prevent infection with SARS-COV-2, prevention or at least reduction of virus transmission in the first place is the most important action to take. Preventative measures, including physical distancing, face covers, hand hygiene and personal protective equipment all aim to reduce the probability of transmission, but can by no means guarantee safety from infection.
Testing in conjunction with contact tracing is another important mechanism to break infection chains by identifying infected people swiftly, and to isolate them and their close contacts. Test and trace strategies vary widely from country to country, with local tracing approaches being the most successful. Current testing procedures for SARS-COV-2 rely on two key features of infection – the virus itself and the immune response by the host. Viral tests check for specific antigens (structures on the outside of the virus) or genetic material of SARS-COV-2 (PCR tests; polymerase chain reaction, referring to the method used to multiply copies of viral genetic material up to a detectable amount in a sample), hence testing for the presence of viral particles during the acute infection (see Figure 20). Antibody tests determine whether specific antibodies which target SARS-COV-2 antigens are present in the host, hence testing whether infection and a subsequent immune response by the host have occurred (see Figure 20).
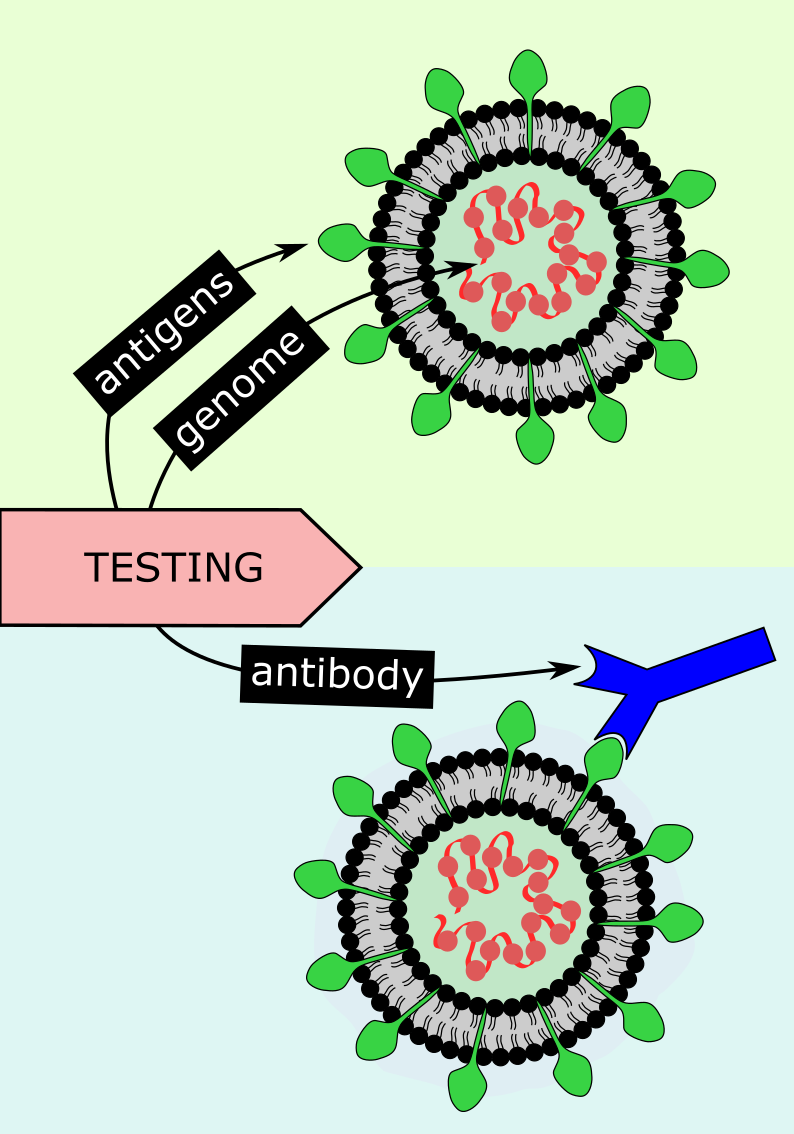
Since antibody testing identifies individuals who have had the virus previously, antibody testing could be used for wider population studies to assess overall transmission patterns of the virus, in retrospect. The accuracy of viral tests is extremely important. Even very small proportions of false positive results may lead to unnecessary isolation of large numbers of people and may nurture subsequent real infection as an individual may wrongly assume they are immune. False negatives can promote spreading of the virus by individuals who believe they are not infected, especially if they experience no or only mild symptoms. An increasingly important issue with PCR testing for acute infection is the interpretation of these tests – whether the detected virus RNA particles are viable (that is, infectious) or not. Overly cautious or overly generous interpretation of these test data are at the heart of false positives and false negatives, respectively. Getting this balance right is by no means trivial but hugely important, for example given the disruptive consequences of extended quarantine periods. Several antigen tests are currently advertised as rapid, potentially DIY / at home test options – this is not likely to become a viable option, however attractive this may sound (antigen tests have a fairly high rate of false positives, are less sensitive than PCR tests, and need to be expertly applied and processed for reliable results). Correct timing of tests is another important consideration in reducing the probability of inaccuracies. For example, antibodies will not be detectable during early infection, and some studies suggest their levels reduce after several months – so two to three weeks post-infection is the ideal time for this type of test.
Safe and effective vaccines are expected to be the gold standard in the prevention of SARS-COV-2 infection and in terminating the COVID-19 pandemic. Vaccines have been a revolutionary medical development, whether in eradicating diseases such as smallpox (the first condition for which this was achieved in 1980), protecting children against serious illnesses (such as polio), or as a precaution when faced with increased risks of a particular infection (such as hepatitis A and B). A successful vaccine would be the ideal long-term solution to the COVID-19 pandemic, providing immunity from infection such that other preventative and interruptive measures are no longer necessary. However, there is no guarantee of a successful vaccine.
In general, vaccines are not a ‘quick fix’ – their production requires a thorough understanding of the virus itself, the identification of theoretically targetable features, and the successful targeting of these features in practise, which often defies theoretical models. Once developed, the safety and efficacy of vaccines must be tested in phases in culture (in the lab), animals and humans. Each step is vital to ensure the vaccine is effective and without hidden adverse effects, but this long process means vaccines can take years to develop and cutting corners in the process poses a safety risk. Safety of any vaccination programme is a prime ethical issue, as healthy individuals are being exposed to a vaccine. Other challenges in vaccine development include viral mutation, which is especially common in RNA viruses such as SARS-COV-2, of which already multiple variants have been identified. The emergence of new strains raises questions for vaccine design, such as how to target multiple strains, and how to prevent re-infection. It could be the case that COVID-19 becomes like influenza (seasonal flu), which requires yearly vaccine adjustment to account for new mutations and strains, with variable efficacy each year. Currently (autumn 2020) it looks like most of the reputable vaccines in advanced clinical trials raise a reasonable immune response, but nobody knows how long this will last and/or how well these vaccines will be able to address clinically relevant mutations of SARS-COV-2 (only few mutations are clinically relevant). At the moment, a best guess might be that these new vaccines will not be as ineffective as any attempts to produce a vaccination against the common cold, probably a little more targeted and robust than the seasonal flu vaccines, but not as useful as hepatitis immunisation and definitely not as effective as vaccinations preventing measles. So probably some repeat patterns of immunisation will be required, and not all vaccines are likely to work equally well for all people.
The goal of vaccination is to trigger a host immune response by using a weakened, partial or artificial form of the virus, such that the immune system will be prepared if infected with the real, wild-type virus. This involves the production of virus-targeting antibodies by immune cells known as B cells, some of which remain in the body (memory cells) and can produce the same specific antibodies if the host is later infected by the wild-type virus. For example, an immune response could be raised against SARS-COV-2-specific spike proteins, after which memory B cells could remain and be able to produce antibodies to target these spike proteins on the wild-type virus. However, the host immune system often has different responses to the vaccine compared to the wild-type virus. The complexity of the immune response likely plays a part in this disparity, with the immune response involving other factors such as T cells (another type of immune cells). Several of these aspects are not yet fully understood in the context of SARS-COV-2 infection and their relevance in the development of severe forms of COVID-19.
Different types of vaccines against SARS-COV-2 are currently (autumn 2020) under development worldwide. The effectiveness of these vaccines is not yet proven. Current advanced, phase III clinical trials include various different types of vaccine (Figure 21):
- non-replicating viral vectors – modified viruses which are unable to replicate, but can carry genetic material of the target virus, and enter relevant cells/tissues where they release viral antigens;
- inactivated viruses – vaccines derived from the natural virus, which has been weakened such that it is no longer infectious or detrimental to the host;
- RNA – RNA coding for viral antigens that enters cells and is transcribed to produce viral antigens (RNA vaccines are a new development, so far none have been approved for clinical use).
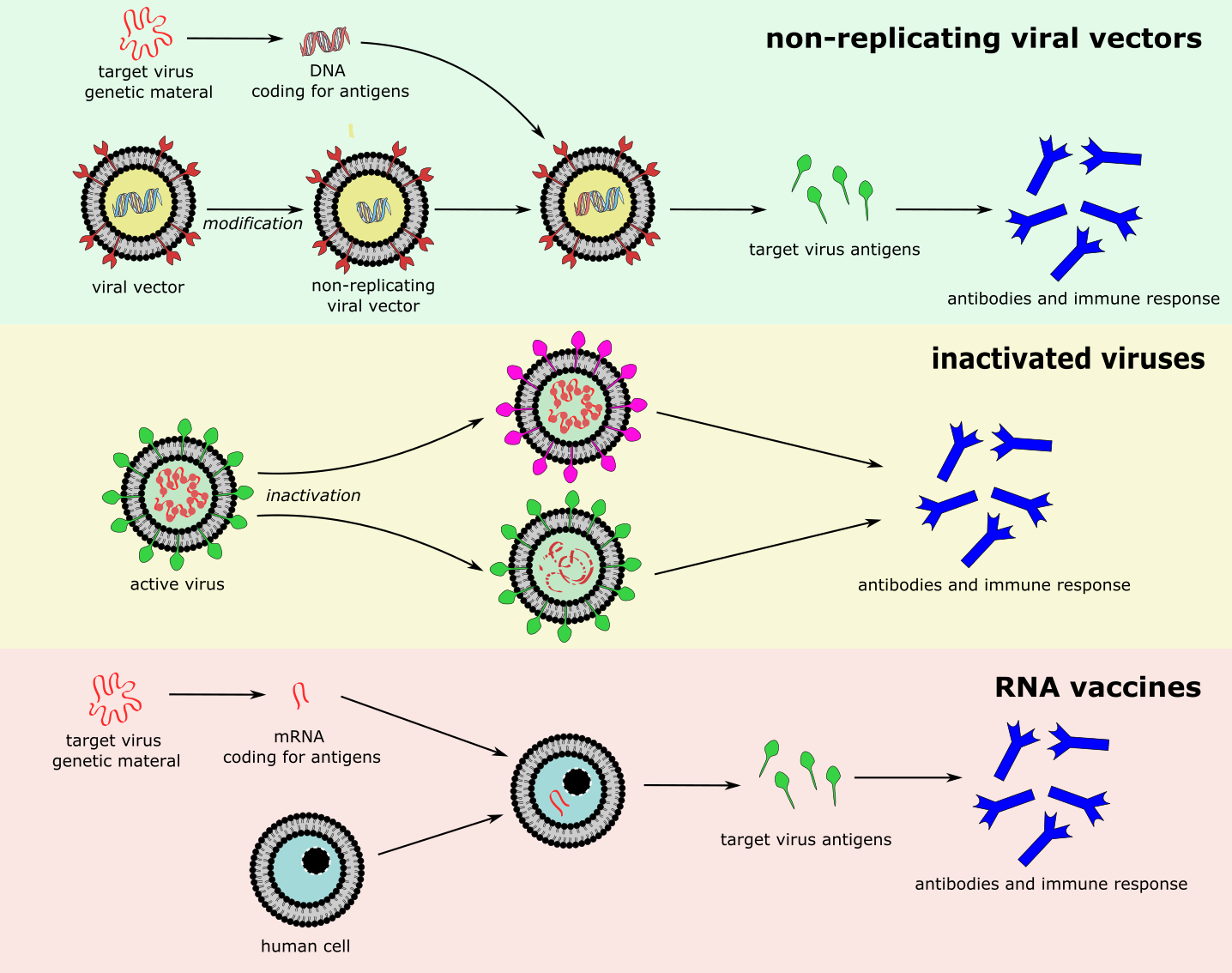
Other vaccines in earlier phases of trials include vaccines based on viral protein subunits, DNA, and replicating viral vectors.
One example of a potential treatment is the much publicised (in the UK) ‘Oxford vaccine’, a non-replicating viral vector in phase III trials. The viral vector is a modified form of an adenovirus that causes common cold in chimpanzees, a well tried & tested vector. The vector carries information into the host cell to synthesise the specific SARS-COV-2 spike protein, which then triggers the immune system of the host to respond by producing antibodies. This approach is a well-established method, in principle. In practice, many detailed aspects remain to be clarified. What will be the strength and duration of any triggered immune response, are there potential adverse effects, short and long term? Will a T cell response also be triggered (assuming this is needed)? Will the response be the same for the wild-type virus as for the vaccine? It is also wise to remember that often, despite best efforts, it is not possible to develop an effective vaccine; for example, even after 30 years there is no vaccination to prevent HIV infection – or, infamously to prevent infection with the common cold virus family.
Especially in the absence of an effective vaccine, it is prudent that other potential treatments are also being explored. For the time being, there is no curative treatment for COVID-19; all that is available are supportive interventions that may (or may not) help the body to fight off the effects of infection when severe forms of COVID-19 develop. Such interventions include
- supply of oxygen, by means of intubation and prolonged mechanical ventilation, or by continuous positive airway pressure (CPAP) via a mask (in the early days of COVID-19 it was assumed it was a pure respiratory pathogen, hence the build of hospitals and accumulation of mechanical ventilators). It is now recognised that it is not a purely respiratory disease. For those who require intubation and mechanical ventilation, there is a 50 % mortality rate (at least in the UK). Non-invasive respiratory support (CPAP) seems to carry a lower risk of mortality (although there are lots of confounding factors in that data) and CPAP creates a fair amount of aerosol);
- anti-inflammatory agents such as dexamethasone and other corticosteroids can have modest beneficial effects in reducing mortality in patients with severe cytokine storm slightly;
- convalescent plasma, obtained from the blood of donors who have recovered from SARS-COV-2 infection, is being trialled. There is no clear evidence about its efficacy, whilst the latest research into the genetics of severe COVID-19 presents findings of auto-antibodies (antibodies targeting the interferon synthesis in the host) in some patients and thus raise issues about the safety of convalescent plasma treatments;
- monoclonal antibodies, laboratory-designed synthetic antibodies that target specific SARS-COV-2 antigens are also being explored; these could be a safer, though expensive backup plan should vaccines and other treatments fail to target COVID-19 effectively.
Update autumn 2021 — The most significant development since autumn 2020 has been the completion of clinical trials and the roll-out of a number of highly effective vaccines against infection with SARS-COV-2. To date, the most successful and most widely used vaccines are those that deliver instructions to the human immune system to produce SARS-COV-2 specific antigens. This has been achieved by either using a viral vector (Figure 21, top; the Oxford/Astra Zeneca, Johnson & Johnson and Sputnik vaccines use this principle) or by packing a small string of viral mRNA in some protective nanolipids to deliver this information/instruction load to the human cells (Figure 21, bottom; the Biontech/Pfizer and Moderna vaccines use this principle). Vaccines using deactivated virus (Figure 21, middle; the Sinovac vaccine uses this principle) seem less effective, based on the information available, whereas vaccines based on specific spike proteins of SARS-COV-2 (the Novavax vaccine uses this principle; currently going through the licensing procedures in several countries) seem to be highly effective, at least in the short term. Overall, the mRNA vaccines achieve the highest levels of protection in very large cohorts of people. All these licensed vaccines are safe, with very small numbers of serious adverse effects and clearly benefits outweigh the risks for the vast majority of people. The vaccines massively reduce severe illness or death from COVID-19 as well as reducing transmission of infection, but asymptomatic or mild disease from infection are not prevented to the same degree. It has also become clear that immunity from both natural infection and vaccination wanes over time, after about six months following complete vaccination the protection deteriorates considerably (more so in older people, starting from a typically lower level of protection anyway, than in young and healthy individuals) and symptomatic, sometimes serious break-through infections in fully vaccinated individuals become more common. There is growing evidence about considerable benefits of a third, so called booster dose, given to people six months after their initial second vaccination dose. From early observations (for example, in Israel), it appears that a booster third dose not only ‘tops up’ immunity in the short term to even higher levels than are typically found after the second dose, but may well be the key to a longer lasting and more robust immune response. It is too early for a definitive view on this latter aspect. There are also practical considerations in terms of vaccine availability and use. The majority of the world’s population has yet to have a first dose but developed countries are administering third doses. While there are pragmatic and ethical reasons why serious consideration should be given to achieving high levels of global immunisation as quickly as possible, the reality is that developed world countries are delivering a third dose to vulnerable groups.
Vaccines using a viral vector as well as mRNA vaccines are very safe but do lead to serious adverse effects in very rare cases. The adverse effects differ for the two types of vaccines. mRNA vaccines have been associated with rare cases of anaphylactic shock (ca. 5 cases per million doses), as well as myocarditis (inflammation of the heart muscle) and pericarditis (inflammation of the lining of the heart) affecting mostly young males (ca. 1 case per million first doses, ca. 5 cases per million second doses); there is typically a swift recovery with no lasting effects.
Adverse effects following vaccination with modified adenovirus as a viral vector are different. Rare cases of Guillain-Barré syndrome (a form of systemic nerve inflammation) have been reported. The most commonly reported serious adverse effect is the serious, potentially life- and limb-threatening clotting disorder, vaccine-induced immune thrombotic thrombocytopenia (the condition is very similar to a rare adverse effect of the commonly used anticoagulant medication heparin). It is essentially a special type of autoimmune reaction, where antibodies that bond with specific surface proteins of platelets (PL4) lead to activation of platelets and the formation of thromboses (clots). Such thromboses affect, for example, the sagittal sinus vein (in the brain) and lead to problems similar to those caused by a stroke. Simultaneously to the formation of thromboses in veins and arteries, there is an enhanced bleeding risk from this radical removal of platelets from the blood stream. Obviously, normal treatment to resolve clots are not a suitable treatment in this situation as this would lead to excessive bleeds. But in line with the autoimmune-reaction character of the condition, high intravenous doses of gamma-globulins help to control and curb the condition, the same treatment that is used to treat the very similar condition that is a rare but serious adverse effect of heparin medication. There are a number of hypotheses about the origin of this adverse effect of adenovirus-based viral vectors in vaccines. Interestingly, it has been known for at least 15 years that such effects do occur when using modified adenovirus preparations as vectors. One explanation could be that tiny amounts of freely ‘floating’ virus DNA in the vaccine preparation not only cause ‘typical’ strong inflammatory reactions to this type of vaccine, but may also be responsible for the formation of the specific antibodies that lead to the formation of platelet clots; it is virtually impossible to completely avoid the presence of very small quantities of virus DNA in the vaccine solution. Another hypothesis is that the adverse effect may be caused by accidental injection of the vaccine into (small) blood vessels. Finally, it has been suggested that part of the problem may be the high dosage of viral vector in these vaccines which, in turn, increases the probability of the presence of very small quantities of adenovirus DNA. The typically fairly strong immediate immune response to this kind of vaccine also points to another potential problem, so called vector immunity. For a second dose, the initial immune response to the viral vector may trigger such an effective immune reaction that some of the second vaccine dose gets eliminated by the immune reaction before it can deliver its information to the body cells (this is the reason why the Russian Sputnik vaccine uses a different viral vector for the second dose). However rare vaccine-induced immune thrombotic thrombocytopenia may be (ca. 10 cases per million doses), it is a serious condition and its high degrees of morbidity and mortality have led to recommendations in many countries not to use this type of vaccine, or to restrict its use to certain demographics. In pragmatic terms it should be noted that one of the companies producing this particular viral-vector vaccine has committed to doing so at cost, unlike all the other vaccine manufacturing companies. There is then a population or global ethical consideration about the use of a life saving vaccine with very low but serious unwanted effects which is being made available in an affordable fashion to the world’s population, against those ten times the cost which are slightly safer and more efficacious.
In more recent developments, promising news have emerged about two oral medications showing good (molnupiravir 50 %) or excellent (paxlovid over 85 %) effects in terms of preventing the development of serious disease or hospitalisation when given early after SARS-COV-2 infection. It is not surprising that design and development of medications takes longer than the design and development of vaccines. It is a considerably more complicated task to get a medicinal drug from the drawing board to clinical use. Both molnupiravir and paxlovid achieve disease suppression by hindering the SARS-COV-2 replication. Molnupiravir increases the rate of SARS-COV-2 RNA mutations, whereas paxlovid hinders a crucial enzyme (protease-3CL) needed for a successful virus replication. Potential adverse effects of both drugs require further careful assessment and observation, but it is thought that protease hindrance as the working mechanism should provide a good safety profile for a drug. Once fully assessed and if successful, oral antiviral medications able to strongly suppress the development of serious COVID-19 may well prove a very important step toward societies being able to manage SARS-COV-2 infections by protecting vulnerable people from serious harm in addition to vaccinations.