Bleeding
Contents
The three main components involved in haemostasis (blood vessel cell walls (endothelial lining), platelets, and clotting factors) all need to work normally and in harmony with each other in order to support normal physiological blood flow and clotting behaviour. Each of the components contributing to these highly regulated mechanisms can fail, or the communication between the components may be faulty. Bleeding disorders can be acquired or inherited (genetic) conditions, which result in some kind of pathological failure of the normal blood flow and/or haemostasis functions. Each component may be overactive, underactive, and/or dysfunctional. Such defects in the coagulation response may cause excessive bleeding from even minor injuries, or, at the other end of the spectrum, blood clots (thromboses) in blood vessels may form inappropriately. Below we sketch the normal function of each of these three main components involved in the various bleeding processes, and give a brief summary of the most common abnormalities and their impact on surgical interventions. We conclude this page with a summary of surgical and pharmacological interventions for the treatment of bleeding after trauma, post-surgical management and common pharmaceutical treatments of bleeding disorders and how these need to be managed ahead of elective surgery.
A wide range of laboratory blood tests play an important role in the diagnosis and monitoring of bleeding disorders and their treatment. These tests range from standard investigations, such as a ‘blood count’, all the way to highly specific investigations into particular clotting factors. For some disorders, especially concerning platelet disorders, the interpretation of blood tests requires special care and may necessitate repeated and/or additional tests to arrive at the correct interpretation.
Normal function and abnormalities of the endothelial layer and cells
Normal function of the endothelial layer and cells
The circulatory system, often referred to as a vascular tree, consists of blood vessels of varying thicknesses, lengths and functions. Throughout the body, blood vessels branch from larger blood vessels to numerous smaller vessels (capillaries) which penetrate the tissues of the body. Blood vessels are made up of different layers, with the thickness of each layer depending on the blood vessel types and functions.
The innermost cellular layer present throughout the whole circulatory system, including the heart and the many different blood vessels, is the endothelial layer (Figure 1). This layer is only one cell thick, and sits between the blood (luminal side; inner volume of the blood vessel) and the basement membrane. Although originally considered to serve only as an inert lining of the blood vessel walls, it has since been understood that the endothelial lining is involved in many different vital functions.
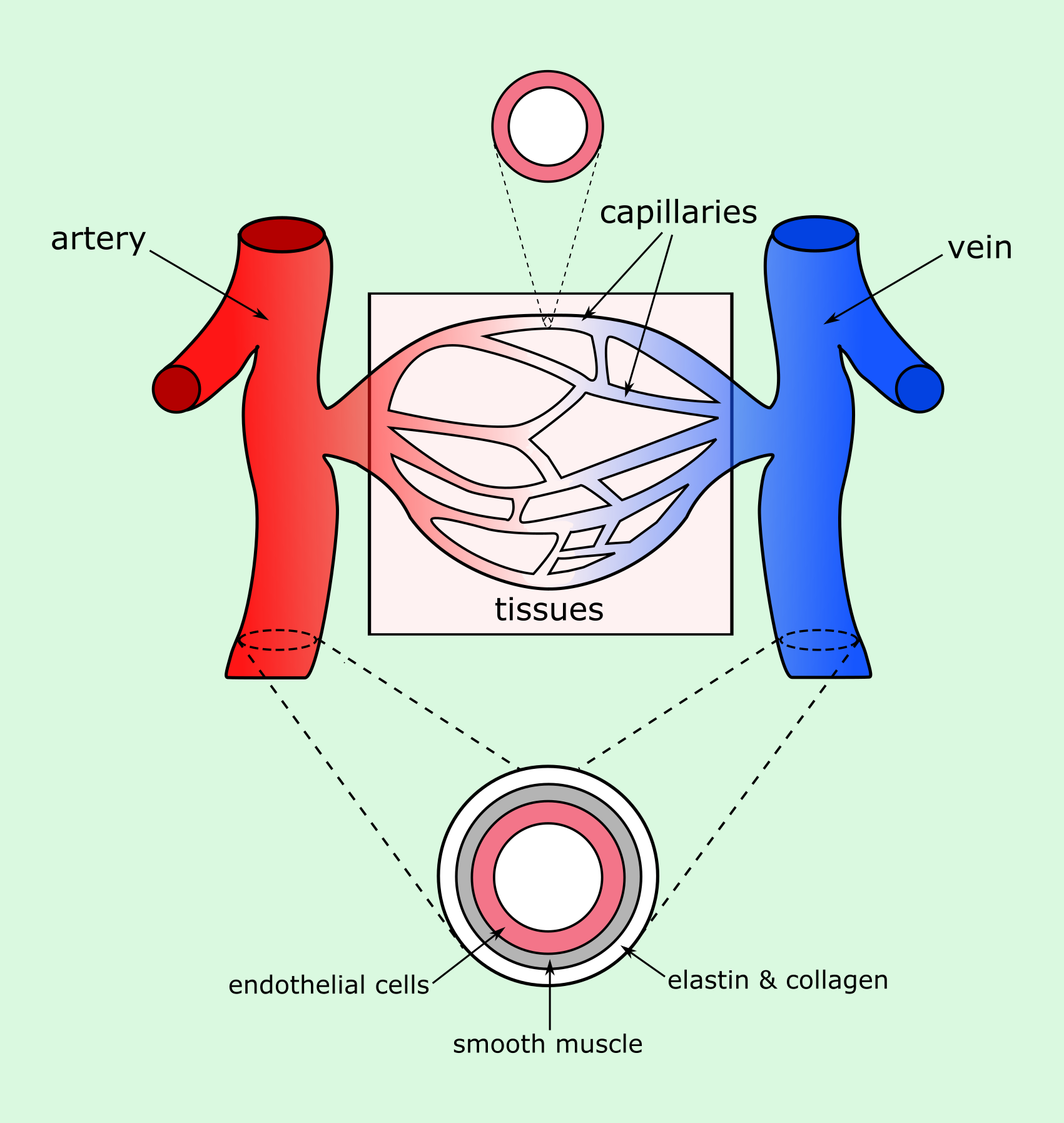
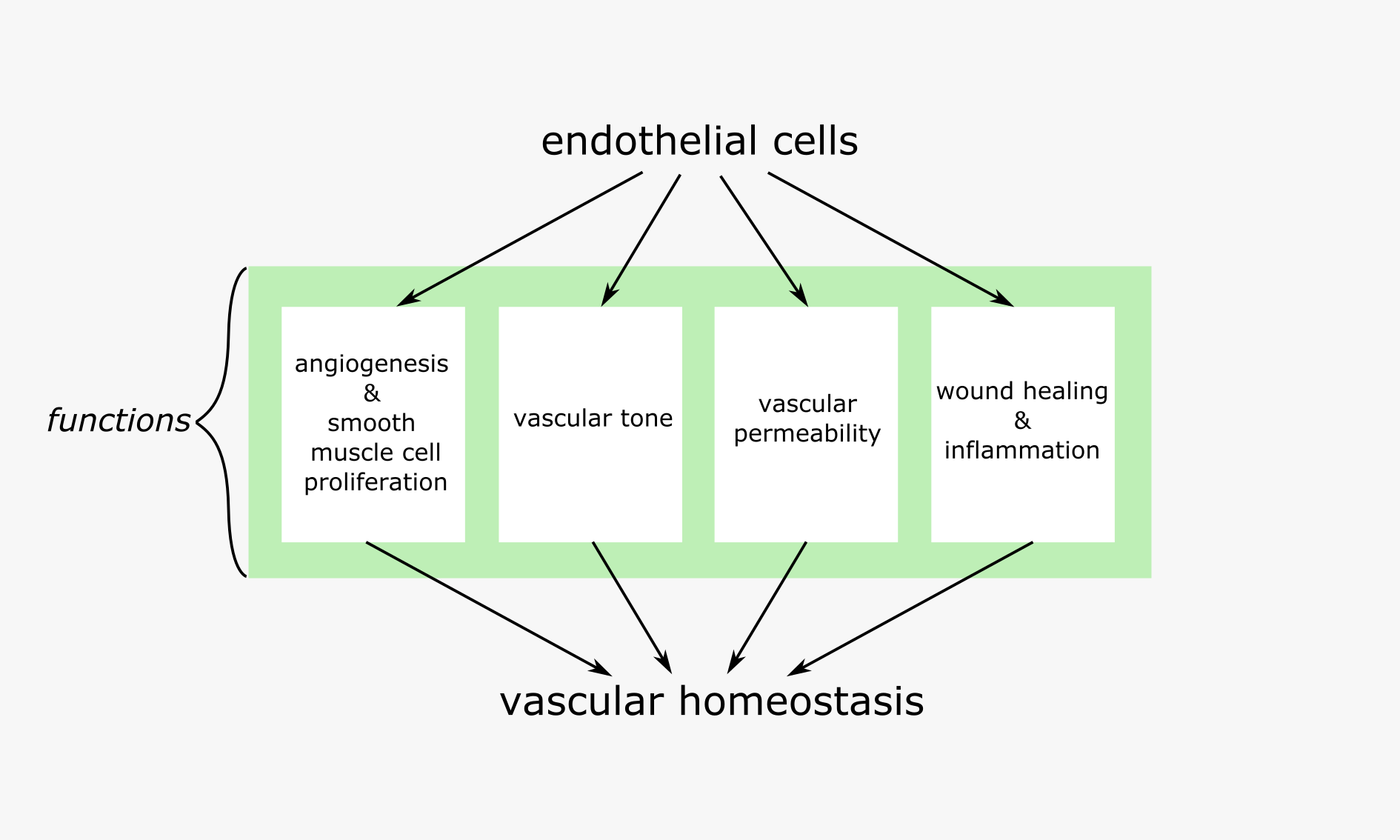
Endothelial cells are responsible for making sure all parts of the vascular tree are working together effectively. They achieve this through a number of homeostatic functions, which centre around maintaining the optimal functionality of the blood vessel network and monitoring the exchange of molecules between the blood and the tissues. In this way the overarching role of the endothelial cells is to ensure the tissues have a rich supply of oxygen and nutrients, whilst preventing the build-up of toxic waste, thus promoting the ideal environment for cellular function.
To meet this need, it is crucial that endothelial cells are adaptive and responsive to their ever-changing surroundings. A number of different receptors on the surface of the endothelial cells allow the cells to constantly monitor and respond to the presence of a variety molecules within their immediate environment (Figure 2). The cells act as a sensor as well as an effector to increase or decrease a range of cellular activities synergistically, releasing and/or signalling the release of regulatory substances as needed to maintain homeostasis.
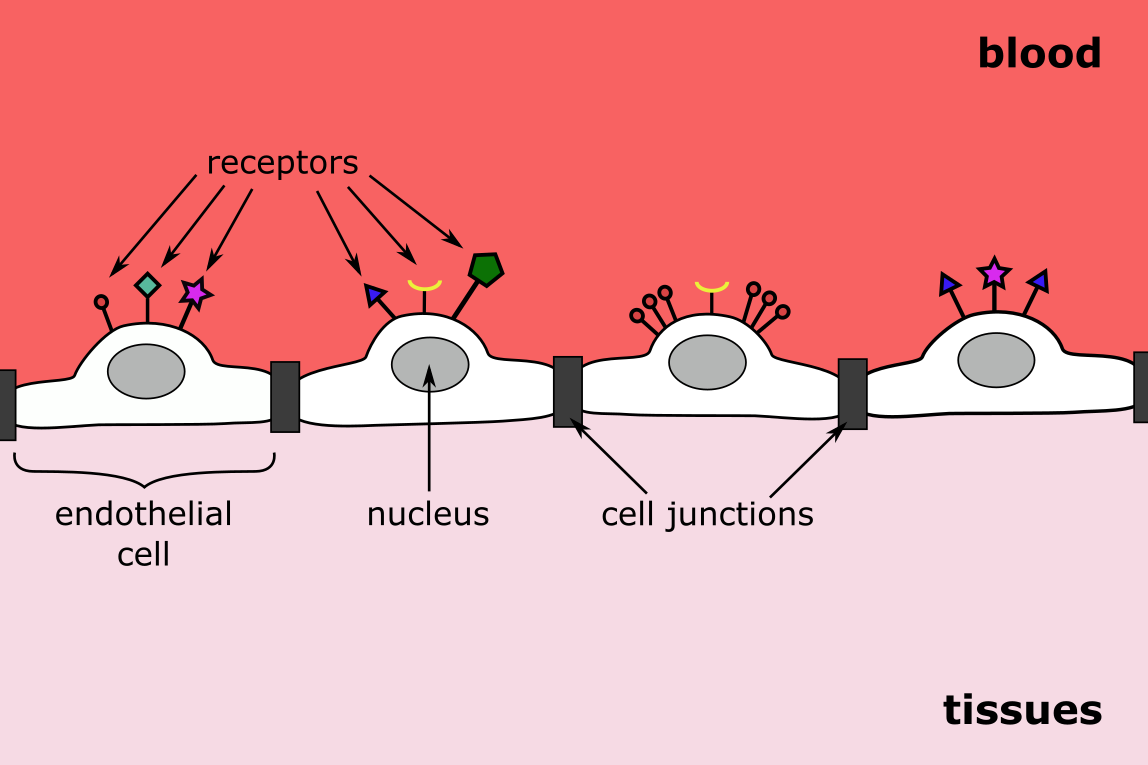
The endothelial layer regulates molecular exchange between blood and tissues, and facilitates wound healing and blood vessel growth throughout the entire vascular network. The needs of specific areas within the network are varied and so endothelial cells are varied in appearance and the extent to which they carry out each of their specific functions. Some tasks are equally necessary at all points of the vascular tree (such as haemostasis), while other tasks at certain points of the blood vessel network can be carried out at different levels. Each and every endothelial cell must be able to adapt to, and be activated by the smallest of environmental changes (Figure 3).
Under normal conditions the endothelial cells constantly produce a range of molecules in controlled amounts. The function of these molecules varies, such as promoting growth or vasodilation, and each molecule often has multiple functions. Without a means to monitor these signalling cascades and regulate them in a rapid-response manner, even small errors in this complex system can result in disease or disease progression. One such molecule of interest is the free radical nitric oxide (NO), which has a key role in a number of functions of the endothelial layer. We use the role of NO in various functions of the endothelium as a ‘worked example’ to illustrate this sophisticated self-regulated system.
Endothelial layer & cells and wound healing
The endothelial cells play an important role in the basic steps of primary and secondary haemostasis (coagulation) and fibrinolysis, necessary for effective wound healing . The endothelial layer helps to promote blood fluidity and makes sure that while haemostasis is promoted in injured areas, blood flow continues as normal in other areas of the vascular network (Figure 4).
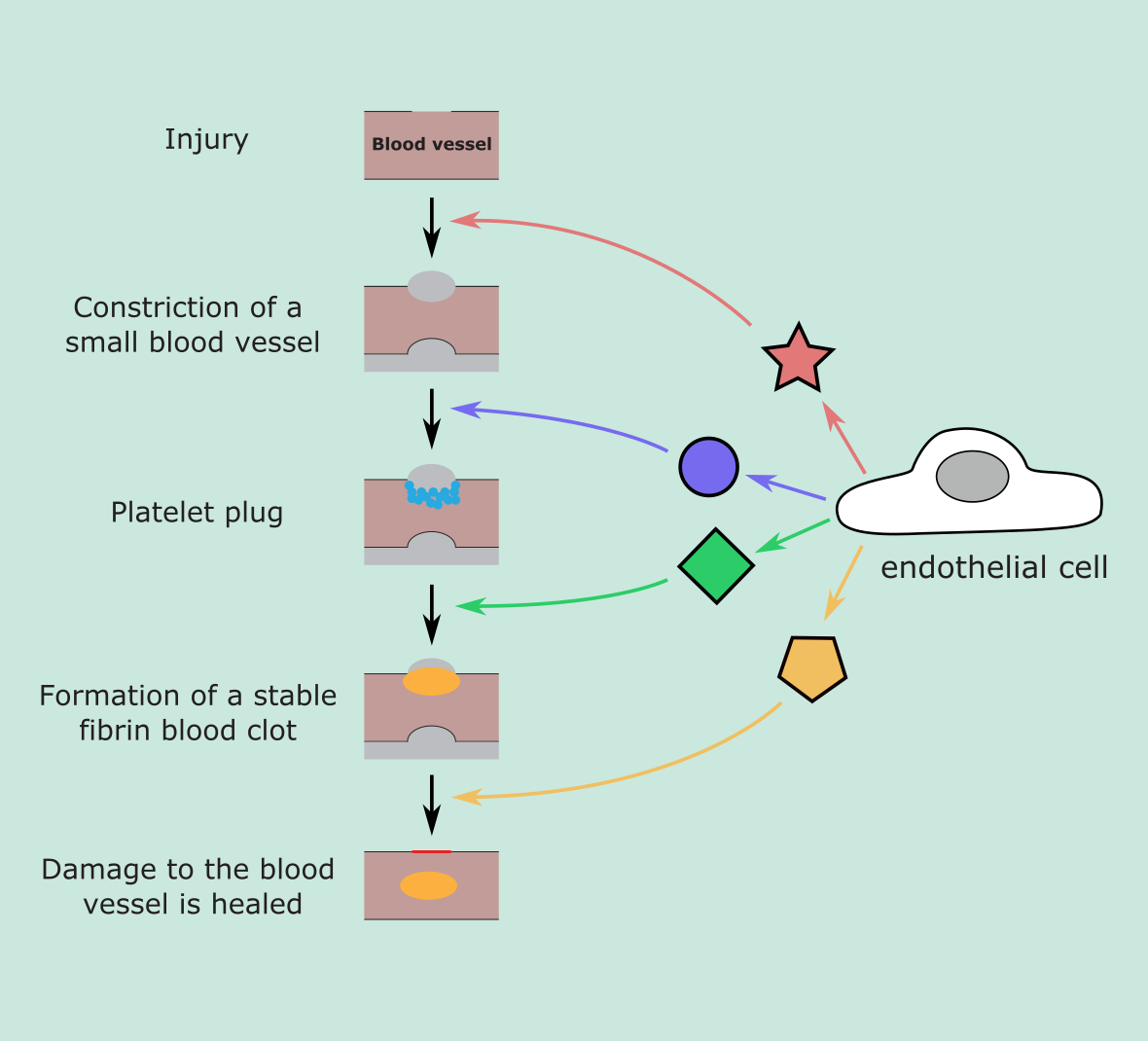
Under physiological conditions, the endothelial cells help to inhibit thrombosis through a number of different ‘antiplatelet’ and ‘anticoagulant’ mechanisms (see below). The non-thrombogenic endothelial layer maintains blood fluidity and controls coagulation mechanisms by adjusting the number of binding sites for anticoagulant and procoagulant factors on the surface of the layer. When a blood vessel is injured, fewer binding sites for inhibitor compounds are presented and a greater number of promoter complex receptors are expressed. Between the blood and the endothelial cells there is layer called the glycocalyx, made up of glycoproteins and proteoglycans. The glycocalyx, amongst other roles, controls the endothelial cell interaction with cells such as white blood cells and platelets. In the absence of injury, white blood cells and platelets are repelled so they do not adhere to the endothelial layer.
The recruitment of platelets and white blood cells to a site of injury in the vascular network is brought about by the release of biological components produced by the endothelium, such as von Willebrand factor (see below) and P-selectin. This initial step in wound healing / stopping bleeding is followed by further wound healing via a complex cascade of interconnected processes, the numerous stages of which are organised through the secretion of different growth factors by endothelial cells.
NO is synthesised during wound healing and plays an important regulatory role in a number of these processes. While the intricacies of the many different pathways in which NO is involved are not fully understood, there is increasing evidence that failure to produce sufficient levels of NO can impact upon effective healing. Under physiological conditions, NO release inhibits platelet activation. Disturbance of this complex balance of molecules involved in pro/anticoagulant systems may result in bleeding or thrombosis. For example, it has been suggested that a build-up of glycated proteins in people with diabetes can promote coagulation in the vascular network.
Endothelial layer & cells and vascular tone
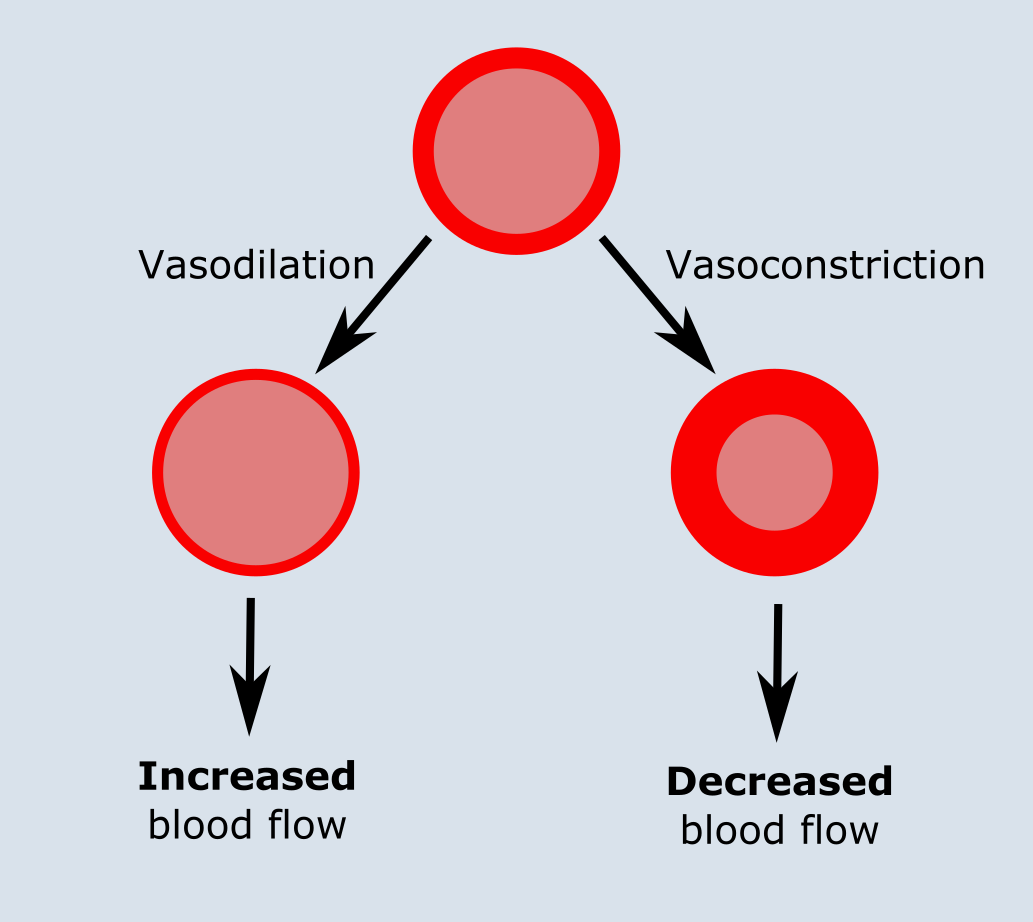
The endothelial layer plays an important role in regulating vascular tone (and indirectly affects blood pressure). It does so through the release of a range of vasodilatory factors and vasoconstrictive factors, such as thromboxane (vasoconstrictor) or prostacyclin (vasodilator). Amongst these factors, NO is a vasodilatory factor which acts on the smooth muscle layer adjacent to the endothelium (Figure 5). NO is released by the endothelium in response to a variety of stimuli including hormones, substances derived from platelets and stress (physical stimuli), resulting in relaxation of the blood vessels and increased blood flow through the vessel. Dysfunction of this endothelium-dependent regulatory system may play a role in cardiovascular diseases, such as hypertension and atherosclerosis (high blood pressure and build-up of plaque in arteries).
Endothelial layer & cells and angiogenesis & smooth muscle cell proliferation
The endothelium and adjacent smooth muscle cells sustain homeostasis of mature blood vessels and are involved in regulating the formation of new blood vessels (angiogenesis; Figure 6), accompanied by the proliferation and migration of vascular smooth muscle cells. These processes are integral for tissue development and repair. In order for effective coordination of blood vessels growth, many different cell types must work together and communicate through inter-cell signalling so that new smooth muscle cells and endothelial cells can be produced to form the layers of the new blood vessels.
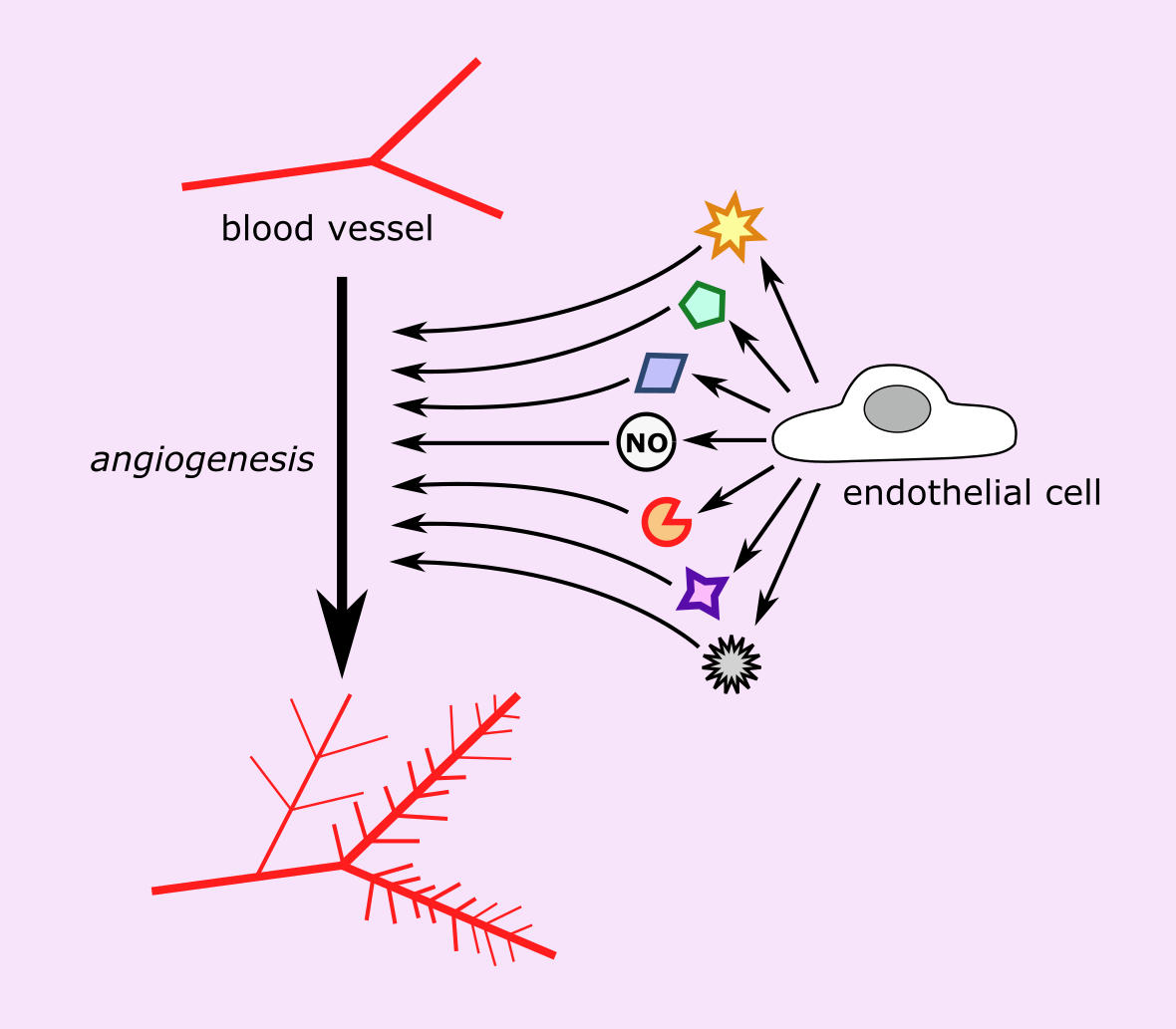
NO produced in the endothelium is crucial to the intricate pathways associated with angiogenesis. NO helps to regulate a variety of angiogenic processes including endothelial cell survival and migration, as during angiogenesis endothelial cells proliferate rapidly and so are less stable. Involvement of NO in angiogenesis and regulating vascular tone are closely related processes.
The endothelial layer also produces molecules to regulate smooth muscle cell proliferation. When a blood vessel is injured, the endothelium releases molecular factors which stimulate the proliferation of new smooth muscle cells. One of the effects of NO is to inhibit smooth muscle cell proliferation, and when the endothelial layer is compromised and not working effectively, less NO is produced. This can result in a loss of control of smooth muscle cell proliferation and consequently lead to vascular disease.
All new tissues, including malignancies, require the growth of new blood vessels to ensure that the tissues are able to exchange vital nutrients with the blood supply. Malignant cells can exploit the physiological mechanisms of angiogenesis by giving off chemical signals which stimulate angiogenesis.
Endothelial layer & cells and vascular permeability
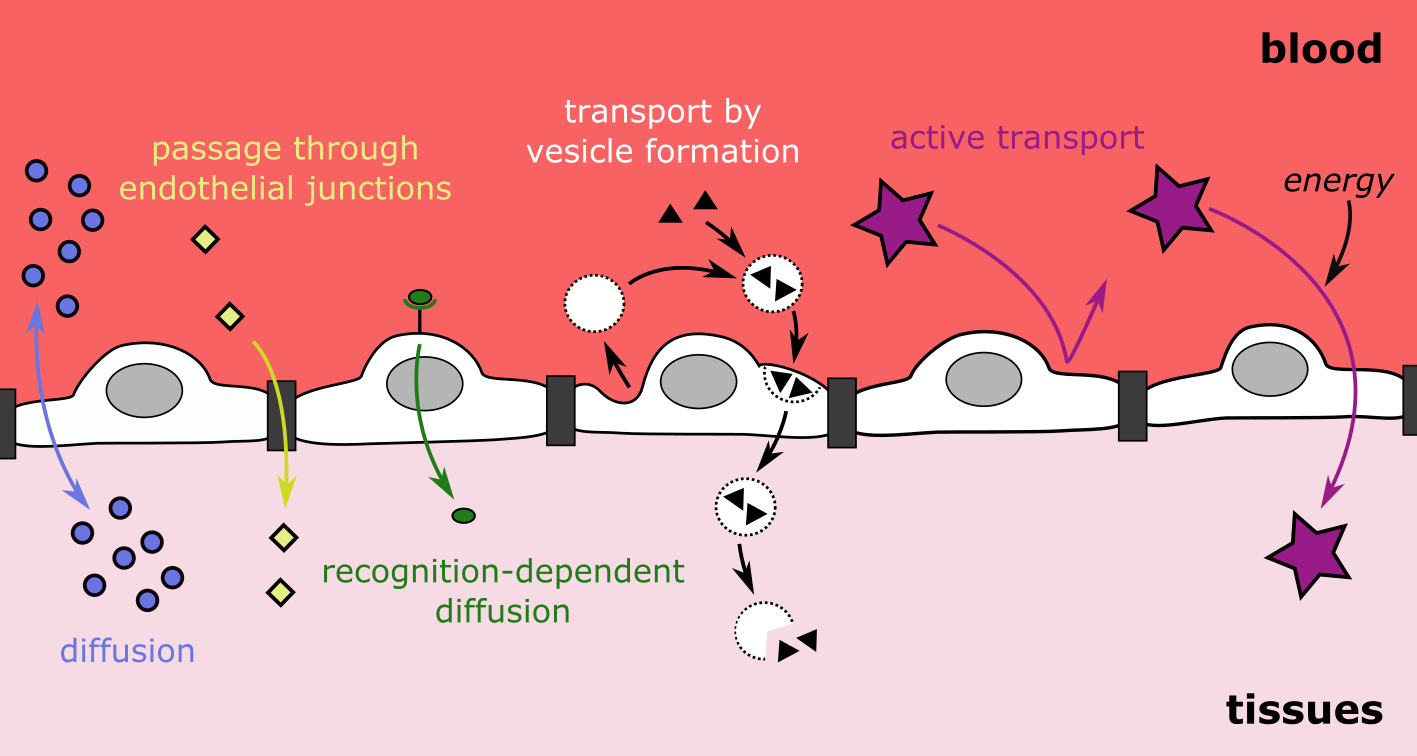
The endothelial layer is a semi-permeable barrier which allows continuous exchange of molecules between blood and tissues. Even at rest, the blood vessels are constantly leaking (extravasation), allowing small molecules such as H2O to pass back and forth across the endothelial layer and actively transporting larger molecules, even cells, to pass across the barrier via channels and vesicles. It is important that solute is able to pass freely between blood and tissues to maintain the interstitial pressure in the various tissues. The endothelial layer contributes to the control of blood flow and the leakiness of the blood vessels through vasodilation and vasoconstriction (see Figure 5) and its effect on blood pressure.
The permeability of the endothelial layer is increased by a number of different protein agents, opening up the junctions between the endothelial cells or causing pores and channels on the surface of the endothelial layer to open also (Figure 7), or by providing energy from chemical reactions.
Free radicals (for example in tobacco smoke ) can cause the endothelial lining to become more permeable and prevent controlled exchange. As a result, the amount of NO produced by the endothelium can become disrupted and imbalanced. Such a breakdown in this synthesis, for example, can result in the establishment and progression of inflammatory conditions , or lead to the development of oedema (collection of excess fluid in tissue).
Endothelial layer & cells and inflammation
When tissues become damaged, the endothelial cells take part in the inflammatory response , a noticeable sign of the of the innate and adaptive immune response mechanisms acting against infection and/or injury.
As the innermost layer of the vascular structure, endothelial cells are crucial to the transport of key molecules and cells to a site of infection and/or damage where the body attempts to eliminate the cause of the problem, resulting in inflammation. During the immune response, the inner endothelial lining becomes adhesive, recruiting white blood cells, and is an enabler of the inflammatory process. Under physiological conditions, NO produced by the endothelial cells prevents the adhesion of white blood cells and platelets involved in wound healing. Dysfunctional endothelial cells are consequently unable to control the immune response appropriately, leading to a response which may be inactive, overactive, or not localised to the site of the damage, which can potentially lead to severe systemic conditions such as sepsis .
Endothelial cell dysfunction
Dysfunction of the numerous roles of the endothelial cells is known to be correlated with an increased risk for heart attacks and stroke. This increased risk is thought to be related to hardened artery walls (atherosclerosis), which can have many different causes, including hypertension, diabetes, high cholesterol levels, older age, or a combination of causes.
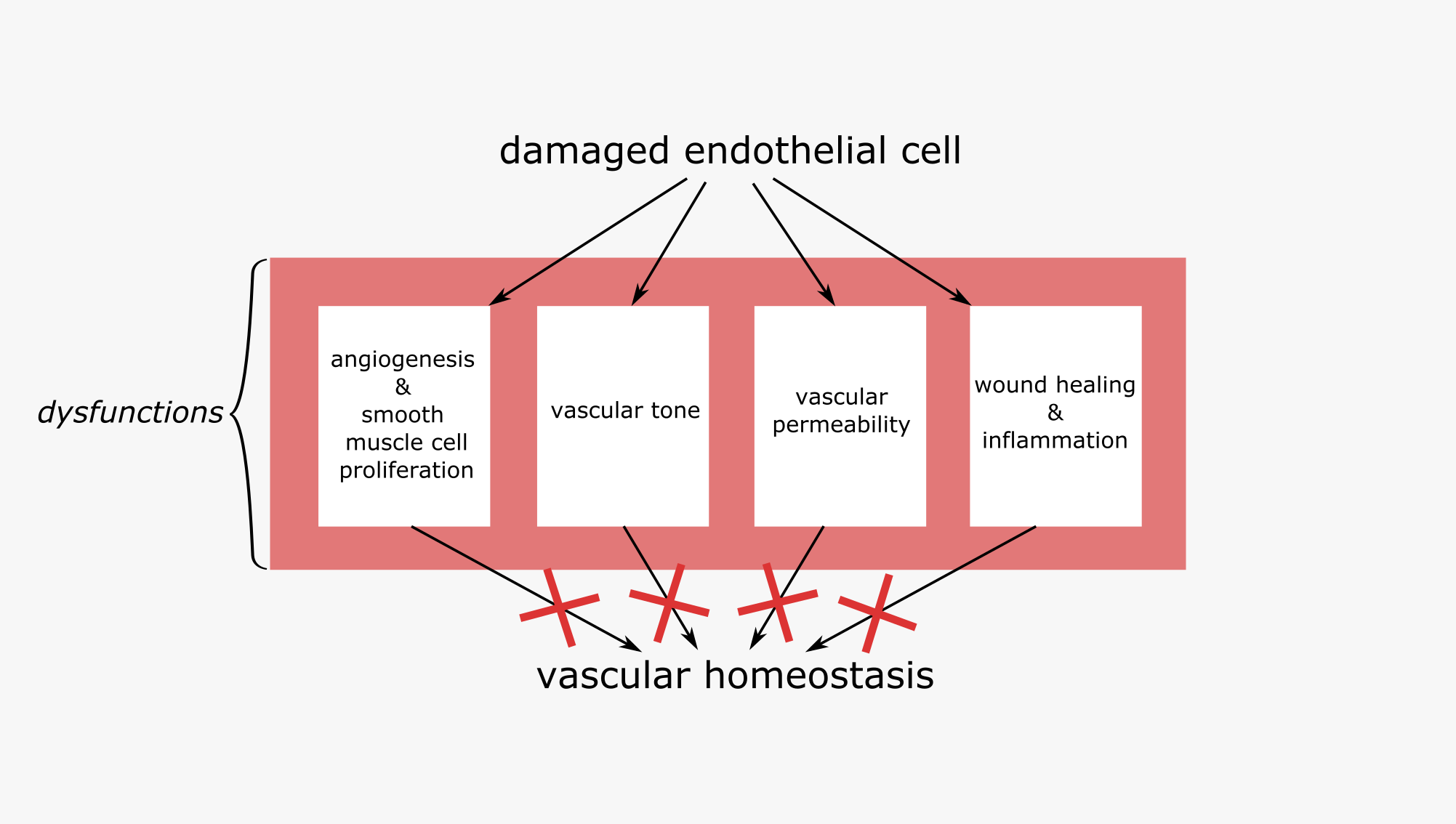
Endothelial dysfunction clearly is relevant for all aspects of bleeding control and wound healing, given the multiple roles of the endothelium in these interconnected processes. Our brief discussion of just a single agent, NO, in the tightly regulated and balanced functions of the endothelium is a reminder that minor imbalances can have major negative effects (Figure 8).
Normal function and abnormalities of the platelets
Clearly, dividing this section into the three main components involved in blood flow and bleeding regulation is a little artificial and oversimplified, given that all the components not only need to function individually but also in a cooperative manner, with considerable overlap of functions and roles. Nevertheless, while keeping this bigger picture in mind, it is helpful to focus on one component at a time.
Normal platelet function
Continuous replenishment of platelets is necessary for the normal function of blood (and bleeding control). The synthesis routes of platelets (thrombopoiesis) thus provide some insight into their functions and failures.
The synthesis of platelets, like the synthesis of other blood cells, involves a process of differentiation from haematopoietic stem cells in the bone marrow. Through a series of progenitor (precursor) cells, these stem cells differentiate into platelet-forming cells known as megakaryocytes (Figure 9). Megakaryocytes mature through replication of DNA and production of proteins, such that the platelets produced have all the material required for their functions. Once mature, these large cells extend branches (known as proplatelets) into small blood vessels, which proceed to release individual platelets from their tips.
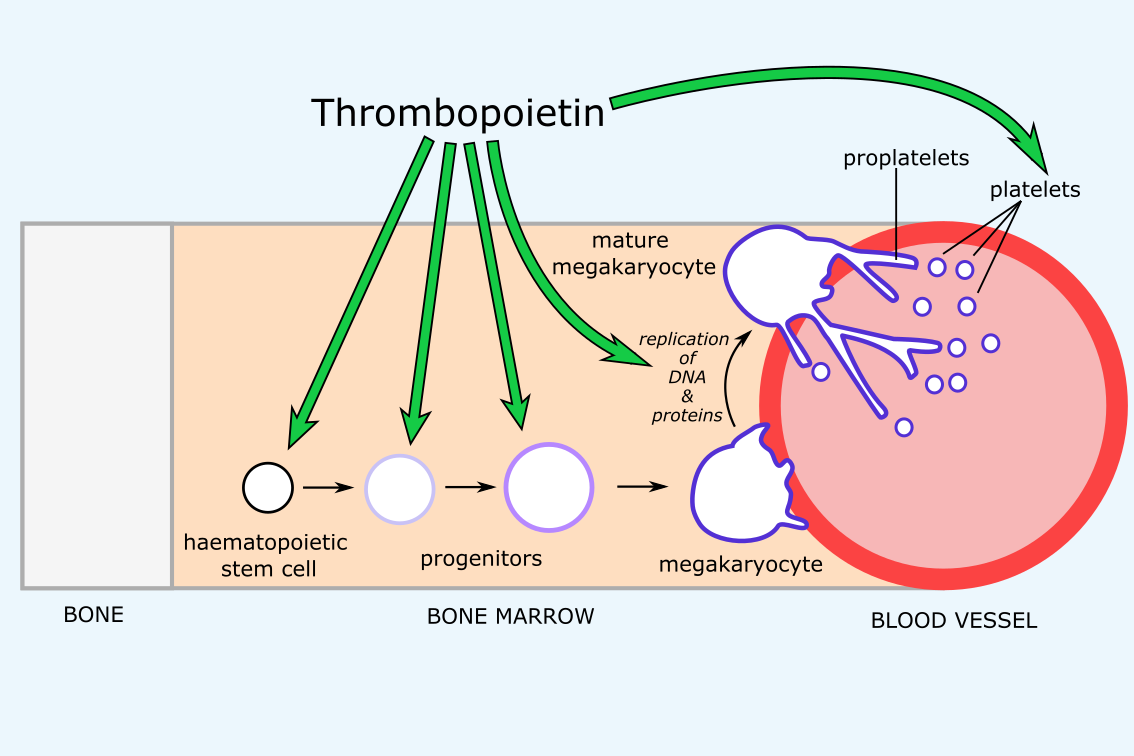
The key regulator of platelet synthesis is thrombopoietin, a cytokine (messenger molecule) produced in the liver, which potentially affects all of the stages of platelet synthesis. Thrombopoietin is thought to promote survival and expansion of haematopoietic stem cells and megakaryocyte progenitors, to direct the formation of megakaryocytes from haematopoietic stem cells, to stimulate the maturation of megakaryocytes, and to promote platelet production and activity. In healthy cells, binding of thrombopoietin to its receptor (the c-MpI receptor) stimulates complex signalling pathways which results in promotion of proliferation and prevention of apoptosis (programmed cell death) of megakaryocytes. One of the key pathways triggered by thrombopoietin is a JAK-STAT signalling pathway (see Figure 10).
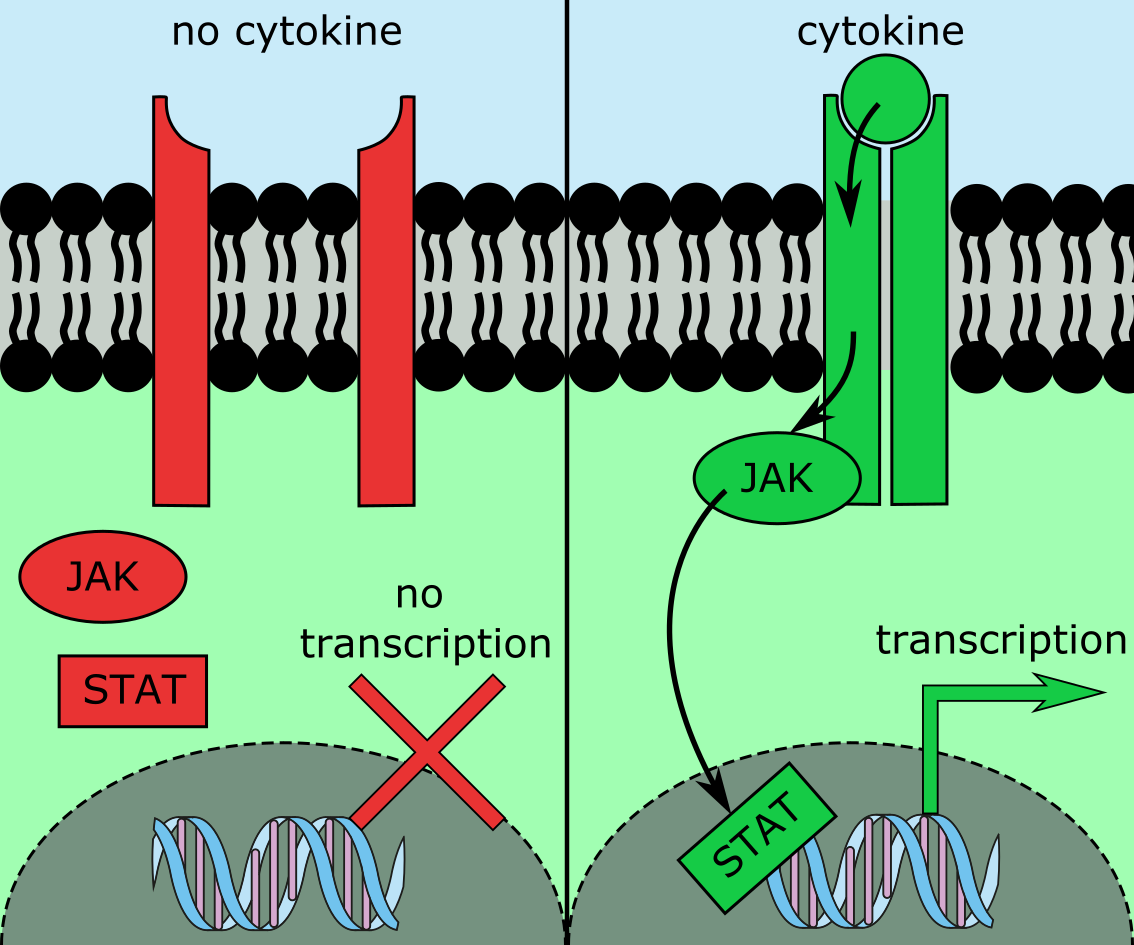
Generally, in a JAK-STAT pathway, Janus kinases (JAK; kinases are enzymes that catalyse specific chemical reactions) are triggered by the binding of a particular receptor to its corresponding ligand, and then activate signal transducer and activator of transcription (STAT) proteins. STAT proteins then act as transcription factors, binding DNA to enhance transcription of certain genes (Figure 10). Such a JAK-STAT pathway involved in the synthesis of platelets is initiated by the binding of thrombopoietin to its cytokine receptor c-MpI, and corresponding effects on proliferation and apoptosis of megakaryocytes.
Normal platelet function involves aspects of adhesion, aggregation and secretions. After injury, platelet adhesion to the endothelial cells lining the damaged blood vessels is mediated by receptors in platelets which bind specific components. In the absence of injury, platelets are not exposed to these components. Initial binding of platelets to the endothelial lining after injury (known as tethering) relies on the von Willebrand factor (see above), a protein which binds to several receptors on platelets that are involved in platelet adhesion. The von Willebrand protein forms bundles anchored in the endothelial cells following damage-related signals after injury. This provides an initial adhesive surface for the platelets, which helps to slow platelet movement and keep them in close proximity to the endothelium, but does not provide stable binding. Following tethering, several collagen receptors bind to exposed collagen and together help stabilise platelet adhesion to the surface. Finally, platelet receptors for other extracellular matrix components such as fibronectins (a type of glycoprotein that promotes cell adhesion and migration) and laminins (another group of glycoproteins, in the extracellular matrix, that are involved in cell adhesion and tissue-structure building) bind to their respective ligands, further stabilising binding.
This stable initial binding (the first ‘emergency response’ ) facilitates signalling pathways to activate platelet integrins (a family of transmembrane proteins that enable adhesion of cells to each other and to their substrates) and change platelet shape.
Activation of platelets is mediated by both adhesion receptors (such as von Willebrand factor and collagen receptors) and soluble agonists (a range of substances that activate responses, such as ADP or thrombin (an enzyme that is necessary in the blood clotting process)). Agonists can be released by either platelets or the surrounding cells. ADP is released by both platelet and red blood cells at a site of injury. Thrombin, the most potent activator of platelet aggregation, is not a direct secretion. Its inactivated form, prothrombin, circulates in the blood under normal conditions, but in the case of injury is rapidly converted to thrombin by factors released by platelets. The various agonists effect a number of characteristic changes associated with platelet activation, including
- change of platelet shape (to optimise interaction with other platelets and the endothelium);
- protein phosphorylation (to promote signalling pathways for activation of platelet functions);
- release of granules containing ligands such as von Willebrand factor or fibrinogen, or agonists such as ADP; these granules promote platelet aggregation;
- activation of the integrin αIIbβ3.
Activation of αIIbβ3 is considered the most important part of platelet activation and aggregation at a site of injury. αIIbβ3 is triggered by agonists and by signalling from von Willebrand factor during platelet adhesion. The symmetry of fibrinogen (a protein forming blood clots; a ligand for αIIbβ3) allows binding to each end of the same ligand by integrins from two different platelets, bringing the platelets into close proximity. Since platelets carry thousands of copies of this integrin, platelets can be brought together on all sides to enhance aggregation. The integrin αIIbβ3 also plays a role in platelet function and regulation, through inside-out and outside-in signalling . All αIIbβ3 action is reliant on an agonist-mediated change of integrin shape, which greatly increases the binding strength between the integrin and its ligands. In a process reliant on thrombin, fibrinogen is eventually converted into fibrin, which polymerises to form a mesh to which platelets bind, allowing formation of a more dense, stable clot (see below). von Willebrand factor can also promote platelet aggregation, by forming multimers which have multiple copies of von Willebrand factor and can hence bind multiple platelets (mutations affecting von Willebrand factor are the most common inherited cause of platelet disorders).
Platelet abnormalities
Platelet abnormalities occur for a broad array of reasons, ranging from various acquired conditions or effects of medications, to autoimmune conditions, and malignancies. These abnormalities may involve a low blood platelet count (thrombocytopenia), a high blood platelet count (thrombocythemia) or changes in the ability of platelets to perform their function (even within normal platelet count range (thrombasthenia)). Dysfunction and inhibition of platelets is most commonly caused by a range of medications (see below) and may be a desired or an adverse effect. Here we focus on thrombocytopenia and thrombocythemia.
A low platelet count, thrombocytopenia, can be caused by changes in platelet production, distribution or destruction (a summary is given in Figure 11).
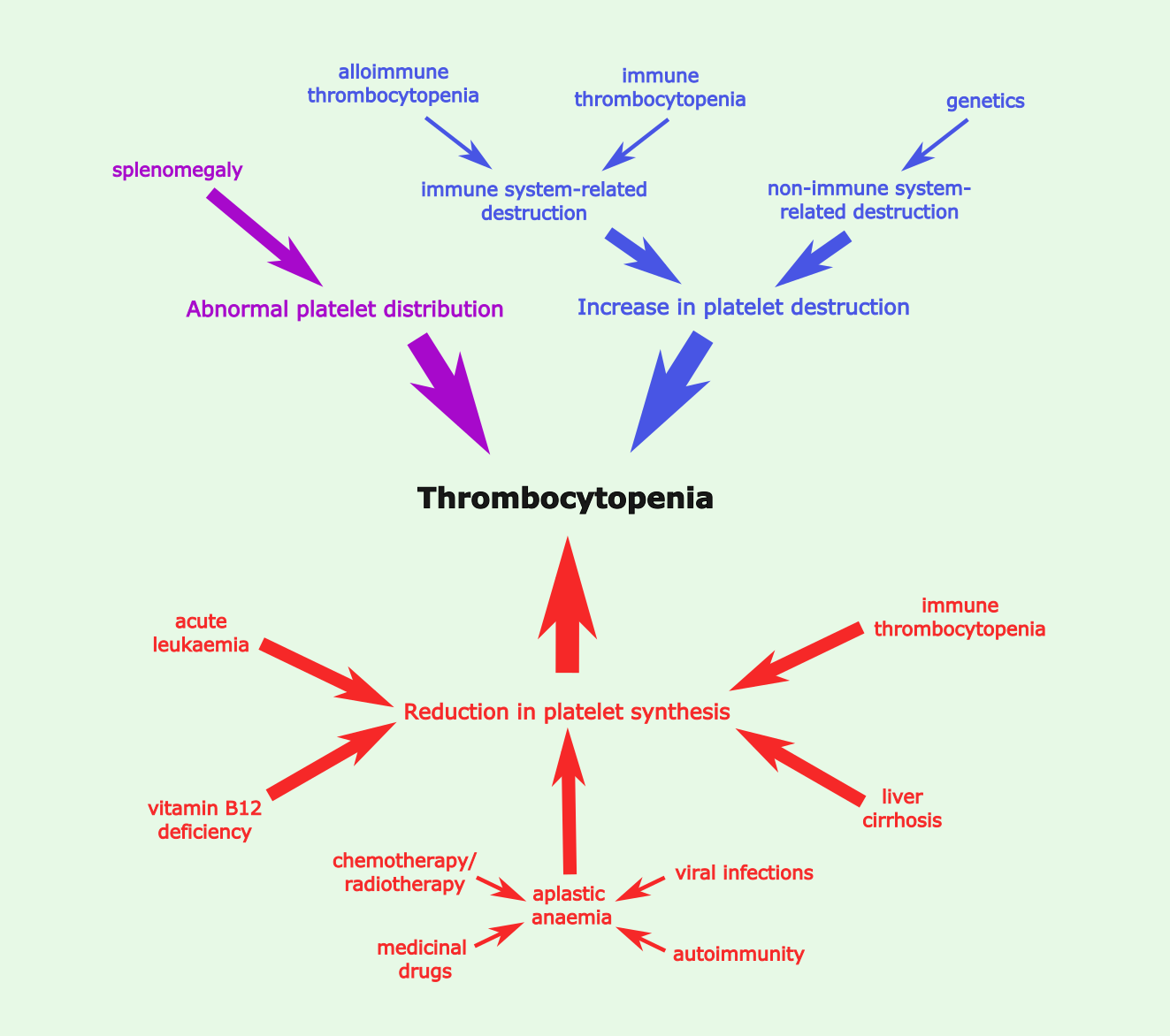
Platelet destruction, and subsequent thrombocytopenia, has both immune and non-immune system-related causes. Immune system-related causes include immune thrombocytopenia and alloimmune thrombocytopenia. Immune thrombocytopenia involves the host immune system attacking its own platelets through antiplatelet antibodies (marking platelets for destruction and their removal by the spleen) and cytotoxic T cells (which destroy the platelets). This can develop on its own (primary immune thrombocytopenia) or less commonly can be associated with underlying conditions which weaken the immune system and/or lead to self-targeting (such as HIV infection and some autoimmune conditions). Antibodies can also play a role in reducing the platelet count in newborns. In this condition (alloimmune thrombocytopenia), a breach of the placental barrier (for example, caused by maternal trauma) results in the mother’s immune system detecting paternally derived platelet antigens, and hence producing antibodies against these ‘non-self’ antigens. Non-immune system-related causes of platelet destruction include inherited thrombocytopenia, such as the Bernard-Soulier syndrome where a defect in the von Willebrand factor receptor complex causes a reduced platelet count and increased platelet size.
Numerous conditions can trigger a reduction of platelet production, and resultant thrombocytopenia. Some forms of blood malignancies (such as acute myeloid leukaemia and acute lymphoid leukaemia) reduce platelet count by excessive production of non-platelet blood cells: acute myeloid leukaemia is characterised by an increase of myeloblasts; whilst acute lymphoid leukaemia is characterised by an increase in lymphoblasts. These immature blood cells and the non-platelet, often dysfunctional blood cells accumulate and take up room in the bone marrow, leaving less room for platelet-producing cells, and hence reducing blood platelet count. Aplastic anaemia can also reduce platelet production, here not by diversion but by destruction of haematopoietic stem cells. This slows or stops production of all blood cells, including platelets (see Figure 9). Causes of aplastic anaemia range from medical interventions including a range of medications (see below), radiotherapy and chemotherapy , to medical conditions including pregnancy, autoimmune conditions where the host immune system attacks its own bone marrow stem cells, and some viral infections (potentially because a virus has similar surface proteins to the stem cells, so antibodies have off-target effects). Other conditions reducing platelet production include vitamin B12 deficiency (vitamin B12 is a co-factor required for the synthesis of platelets and other blood cells; surprisingly common deficiency with the advent of proton-pump inhibitors (antacid medications) and fad diets), liver cirrhosis (where scarring of the liver causes reduction of thrombopoietin and hence platelet production – see Figure 9), and some forms of immune thrombocytopenia, where antiplatelet antibodies are thought to impair the formation of megakaryocytes.
Abnormal platelet distribution can also have an effect on blood platelet count. For example, disorders which cause splenomegaly (an enlarged spleen) can cause increased sequestration of platelets in the spleen, thus reducing platelet count in the blood circulation. However, this is rarely a cause of bleeding, as in stress conditions sequestered platelets are released from the spleen.
These illustrative examples demonstrate the complexity of the origin of thrombocytopenia: many conditions can cause a reduced platelet count; many of these conditions have numerous causes themselves. There are many more conditions affecting platelet count, but these have not been mentioned here. Thrombocytopenia and underlying conditions are very much interconnected, with underlying conditions both causing thrombocytopenia and occurring as a result of thrombocytopenia. Nevertheless, all of these conditions have in common that the reduced platelet count is associated with excessive bleeding and bruising, and in severe cases can cause life-threatening internal bleeds or brain haemorrhages.
A high platelet count, thrombocythemia, typically results from underlying acquired conditions (reactive thrombocythemia), but can also be inherited (hereditary thrombocythemia), or a direct result of a myeloproliferative disorder (blood malignancy; essential thrombocythemia) affecting stem cells and their proliferation to blood cells.
Reactive (or secondary) thrombocythemia causes approximately 80 % of thrombocythemia cases. The high platelet count is caused by various underlying systemic disorders (for example malignancies, chronic inflammatory diseases, infectious diseases and many others), and/or certain drugs (see below). These conditions can result in increased levels of signalling molecules such as hormones and cytokines, including thrombopoietin, which promote platelet production. Hereditary thrombocythemia can involve some of the same mediators of platelet levels, for example being passed on through inheritance of a dominant mutation of the thrombopoietin gene. The mutation makes the thrombopoietin gene resistant to translation repression, causing overproduction of thrombopoietin and hence platelets.
Essential thrombocythemia increases production of the platelet-forming megakaryocytes. Mutations affecting the JAK-STAT signalling pathway (see Figure 10) are a common observation here; mutations in the c-Mpl receptor, JAK protein or other related factors can all result in a constantly active pathway (even when thrombopoietin is absent), which results in excessive production of platelets by megakaryocytes. Since c-Mpl is also found on platelet surfaces, it normally plays a role in reducing available free thrombopoietin. However, some mutations are also thought to reduce c-Mpl present on platelet surfaces, thus increasing thrombopoietin available to stimulate platelet synthesis (see Figure 9).
All forms of thrombocythemia result in an elevation of the platelet count. This can cause abnormal clotting (see below) and blockage of blood vessels, sometimes resulting in very serious conditions such as heart attacks (blockage of blood flow to the heart) or stroke (blockage of blood flow to the brain). In maxillofacial surgery, such blockages (thromboses) are the most common cause of failure of free flaps in reconstructive surgery. Less commonly, a large excess of platelets can (somewhat counterintuitively) result in excessive bleeding. Disseminated intravascular coagulation, with abnormal clotting in all blood vessels resulting from overactivated clotting mechanisms, is a dreaded complication (or unwanted effect) of massive trauma, massive transfusion, haemorrhage, infection and sepsis. Similar unexpected affects can occur in thrombocytopenia, with low platelets sometimes resulting in increased clotting. A complete dysregulation of platelets and clotting reflects severe systemic conditions such as sepsis and other serious inflammatory conditions.
A number of common medications have significant effects on platelet synthesis and/or function. This may be a desired effect in order to manage platelet disorders (see below), or may occur as an adverse effect of treatments for other conditions.
Adverse effects of common medications on platelets include
- nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen or aspirin – impair platelet function by blocking synthesis of thromboxane A2 (a substance that triggers platelet aggregation). In fact, aspirin in low dose is also used as an ‘antiplatelet agent’ in the prevention of strokes and heart attacks;
- diuretics (agents to treat oedema) – for example, furosemide strongly inhibits platelet aggregation;
- gold-based agents for the treatment of autoimmune conditions such as rheumatoid arthritis – can induce antibodies to platelets;
- antibacterial agents – for example, penicillin impairs platelet function by hindering interaction of agonists with platelet surface;
- antacid medications such as ranitidine or proton-pump inhibitors – reduced platelet counts are repeatedly described in the literature;
- statins (agents to reduce cholesterol levels in blood) – decrease platelet aggregation directly and indirectly (by hindering NO synthase production);
- oestrogen-related medications such as oral contraceptives and hormone-replacement therapy – increase platelet count and risk of blood clots;
- tamoxifen (agent to treat hormone-receptor positive breast cancer after initial treatment to reduce risk of metastasis and recurrence) – directly hinders platelet aggregation. In fact, it has been suggested that this platelet inhibition may be an important factor of the desired effects of tamoxifen in reducing metastasis risk for such tumours (platelets are thought to play a crucial role in the dissemination of malignant cells via the haematogenous route (via blood vessels)).
Abnormalities of coagulation (coagulopathies)
Coagulopathies are disorders that affect secondary haemostasis, where physiologically a weak initial platelet plug is consolidated and strengthened by a cascade of factors. Most coagulopathies are genetic disorders, with only a few belonging to the class of autoimmune disorders or being caused by acquired conditions. Similar to platelet abnormalities, also coagulopathies fall into categories of over- or underactivity.
Following injury, secondary haemostasis encompasses the activation of clotting factors, conversion of prothrombin to thrombin by clotting factor Xa, and conversion of soluble fibrinogen to insoluble fibrin enabled by thrombin during clotting. The clotting process follows complicated parallel pathways (intrinsic and extrinsic pathways with slightly different effects on bleeding pathology) of activation and convergence, before a stable clot is achieved. Calcium ions are necessary for the promotion and acceleration of nearly all clotting factors.
Causes of the most common coagulopathies include
- excess bleeding:
- vitamin K deficiency (can be caused by anticoagulant medications (warfarin) or some antibacterial agents, or by malabsorption of fat (vitamin K is fat-soluble) – prevents the activation of calcium-ion dependent proteins;
- haemophilias – typically genetic deficiency of one or several clotting factors (either insufficient synthesis or poor function), the most common causes of coagulopathies;
- liver disease – an acquired cause for insufficient production of clotting factors (and platelets, see above).
- excess clotting:
- factor V Leiden – a fairly common genetic mutation of factor V gene, usually asymptomatic but in a minority of cases the mutation leads to significant thrombophilia by increased factor V activity; in the literature, factor V Leiden has been associated with the failure of free flaps in reconstructive maxillofacial surgery;
- antithrombin III deficiency – genetic disorder with low levels of antithrombin;
- prothrombin mutation (factor II mutation) – excessive factor II production leads to increased clotting;
- antiphospholipid syndrome – a rare disorder leading to increased clotting by a faulty immune system response and antibody production; can be associated with autoimmune conditions such as Sjögren’s syndrome, some infections such as syphilis or Lyme disease, inherited, or idiopathic (unknown cause).
Surgical and non-surgical methods to treat abnormal bleeding / clotting
Non-surgical methods
Widely used, often long-term medications in the prevention of excessive clotting and related risks for thromboses and other abnormal bleeding conditions include
- excess clotting:
- antiplatelet medications – such as clopidogrel and prasugrel (impair platelet function by inhibiting ADP-induced platelet aggregation) or ticagrelor (inhibits a particular platelet receptor);
- anticoagulant medications – such as heparin (widely used to prevent deep vein thromboses after surgery; heparin causes a drug-induced immune thrombocytopenia) and warfarin (reduces the synthesis of various clotting factors by depleting vitamin K; the effects of warfarin can be quickly counteracted by administration of vitamin K);
- low-dose aspirin – mainly used in the treatment of primary thrombocythemia in the management of high risk for development of blood clots;
- some glycoprotein inhibitors (platelet integrin complexes) strongly inhibit platelet activation and are only used for specific applications such as in angioplasties or for placing stents in coronary blood vessels;
- breaking down blood clots is supported by administration of plasminogen activators such as urokinase or streptokinase (these catalyse the conversion of plasminogen to plasmin; plasmin is essential for fibrinolysis);
- excess bleeding:
- treatment of thrombocytopenia depends on the underlying cause(s) and may include supplementing vitamin B12, giving corticosteroids or other immunosuppressant agents, blood or platelet transfusions;
- various clotting factors have to be administered to manage significant cases of haemophilia (in the UK all haemophiliacs are registered with a centre who can advise on management of trauma or surgery);
- the vasoconstrictor properties of adrenaline help reduce superficial bleeding;
- tranexamic acid binds and inhibits plasminogen, thus preventing fibrinolysis (see below); it is now part of the ATLS trauma bleeding protocol (advanced trauma life support).
Surgical methods
The most common cause of bleeding is trauma, accordingly surgical methods to treat bleeding are fully integrated in trauma surgery – in maxillofacial surgery as well as in other branches of surgery.
Surgical methods to stop bleeding start with compression of the bleeding site and then often involve ligation of vessels, extreme temperatures / diathermy and microsurgical repair of injured blood vessels. Tranexamic acid is an effective means to rapidly stop excessive bleeds after trauma. Ongoing medication with antiplatelet or anticoagulant medications can add considerably to the burden and impact of trauma by exacerbating bleeding.
The situation is different for elective surgery where a managed approach to reduce or stop antiplatelet or anticoagulant medications ahead of surgical intervention is a common approach. When major blood loss is to be expected in elective surgery, sometimes well timed own-blood donation prior to surgery is an option to avoid donated transfusion reserves.
Bleeding and bleeding disorders are immensely important considerations before, during, and after surgical intervention – not least because of profound effects on post-operative timelines and outcomes.