Pain
Contents
Across the world, the general number 1 reason to seek medical support is pain. In 2019 analgesics are the most widely used of all medicinal drugs, worldwide. No wonder that nowadays there is a bewildering range of analgesic agents (and other approaches to manage pain). Adequate treatment of pain, a wider recognition that acute and chronic / neuropathic pain are very different circumstances, as well as an increasingly more honest attitude toward adverse effects, has been a complicated journey over the past decades.
In 1986 the World Health Organisation (WHO) proposed a simple scheme (which became known as the WHO analgesic ladder; Table 1) of escalating pain medication according to need, in order to improve the analgesia provision for cancer patients who often suffered unnecessarily from severe and undertreated pain. It was recognised that some irrational fears had to be overcome: fears by clinicians to prescribe opioids, and fears by patients to use opioids. The scheme certainly helped in that respect, but over the decades it has become outdated as more and different analgesics and delivery options were developed. Some refinements and expansions were added to the simple original scheme over time.
Table 1 The WHO analgesic ladder as of 1986, for the treatment of cancer-related pain
| Pain characteristics | Suggested medication scheme |
|---|---|
| step 1: mild pain | paracetamol and NSAIDs |
| step 2: moderate pain | paracetamol and NSAIDs, plus weak opioid |
| step 3: severe pain | paracetamol and NSAIDs, plus potent opioids |
However, it seems sometimes to have been forgotten that the scheme in Table 1 was never meant as a guide to treat chronic / neuropathic pain conditions. Especially in North America this oversight, in combination with powerful (if not dubious) marketing methods by the pharmaceutical industry, lead to what now has become a full blown epidemic of opioid use, with all the concomitant personal and societal adverse effects one should expect in that situation. Fortunately, the European approaches to pain control seem to have been more thoughtful and temperate. However, prescriptions of opioids have also increased in Europe over recent years, and there are reasons to believe that opioids are increasingly and inappropriately prescribed for the management of chronic / neuropathic pain conditions. Over the past decade more appropriate schemes for the management of chronic / neuropathic pain have been developed. And yet, it strikes one as a sad fact, if in 2016 the British Medical Journal sees a need to publish an editorial reminder, in no uncertain terms, about failures past and present in the use of analgesic drugs for the management of chronic / neuropathic pain.
Below we give an overview of analgesics used in the management of oral and maxillofacial conditions. Our discussion groups analgesics together according to their mechanisms of action.
Non-steroidal anti-inflammatory drugs (NSAIDs)
Mechanism
Non-steroidal anti-inflammatory drugs (NSAIDs) are organic chemicals, most of them carboxylic acids. NSAIDs have analgesic, antipyretic (prevent or reduce fever) and anti-inflammatory effects. They act in different places in different circumstances, often depending on the mode of delivery of the drug, but generally act in the periphery by reducing hyperalgesia.
Non-steroidal anti-inflammatory drugs act by inhibiting the family of cyclooxygenase (COX) enzymes by blocking the catalytic site or channel of these enzymes (Figure 1). There are two closely related COX enzymes, COX-1 and COX-2. COX-1 supports the ongoing synthesis of the various prostaglandins needed for general, normal cell functions, including maintenance and protection of the lining of the gastrointestinal tract. COX-2 is involved in the synthesis of most of the prostaglandins and thromboxanes contributing to inflammation, nociceptive sensitisation (enhanced pain perception] and fever.
Blocking or inhibiting COX enzymes thus inhibits the production of prostaglandins and thromboxanes, which is thought to be the mechanism leading to the dominant analgesic effect of NSAIDs. Non-steroidal anti-inflammatory drugs inhibit both COX enzymes, so also inhibit processes needed for general, normal cell functions which explains their well known and widely experienced adverse effects (see below).
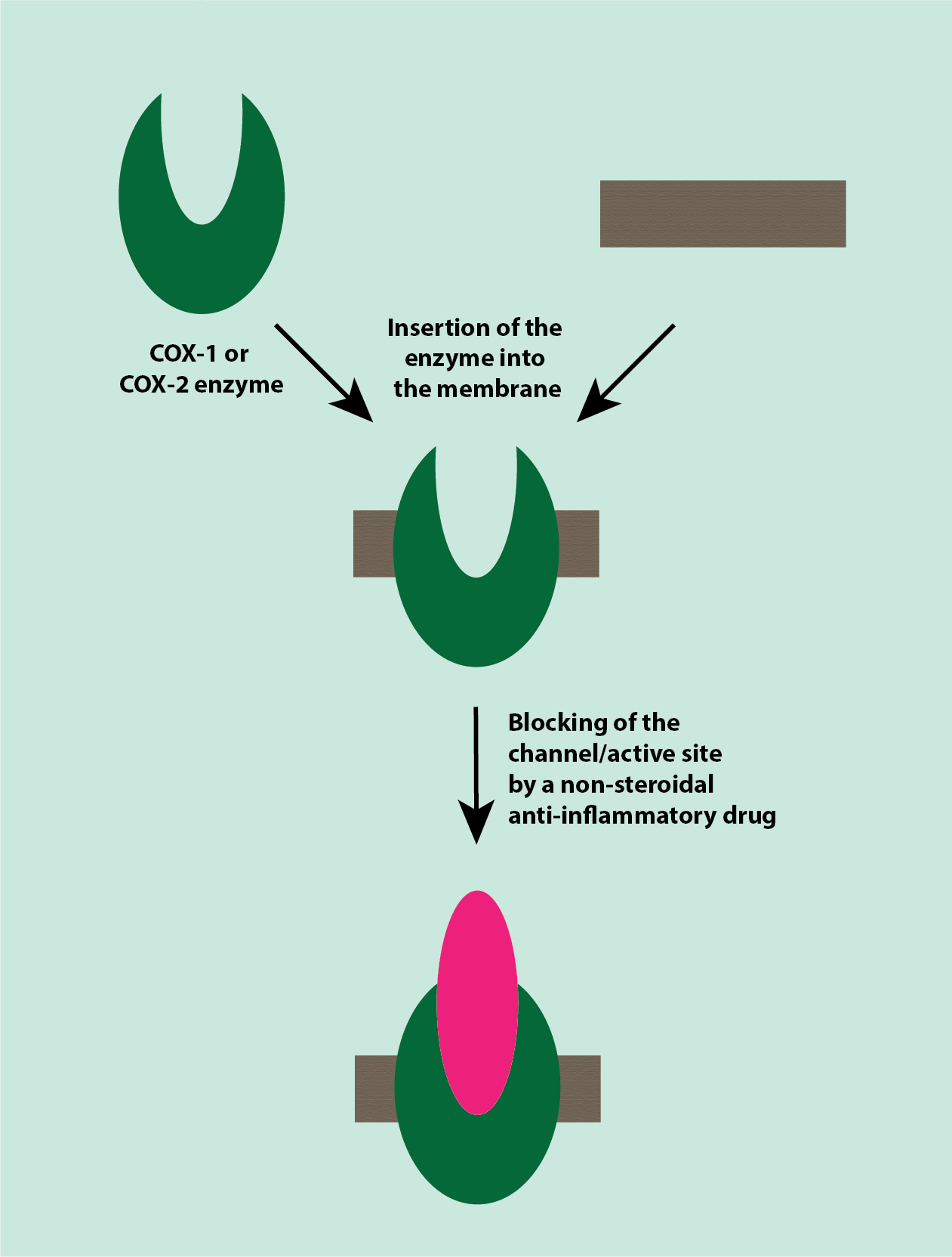
Non-steroidal anti-inflammatory drugs attenuate abnormal pain responses in inflammatory conditions, they have no obvious effect on normal pain thresholds and only work to overcome mild to moderate pain in inflammation.
Aspirin is a non-steroidal anti-inflammatory drug with additional functions (suppressing normal platelet functions in blood clotting), beyond its analgesic role. Unlike other NSAIDs, aspirin has long-lasting inhibitory action on the COX enzymes.
List of NSAIDs commonly used in maxillofacial surgery
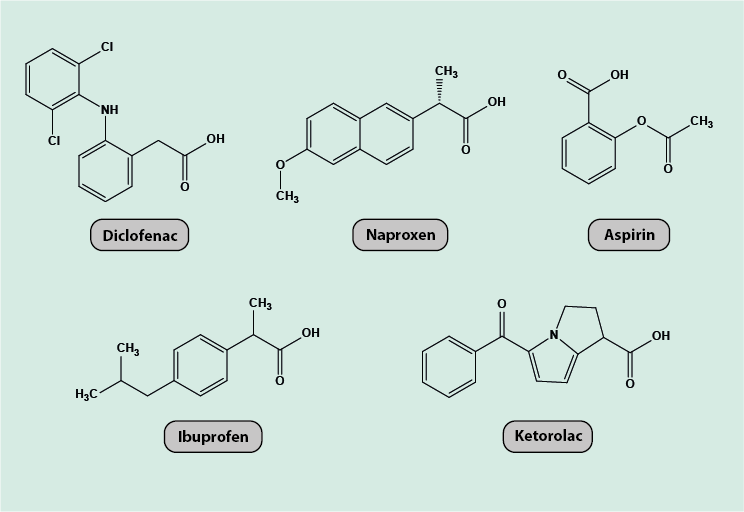
Figure 2 depicts the chemical molecular structures of five commonly used NSAIDs, diclofenac, naproxen, ketorolac, ibuprofen and aspirin; all of which are carboxylic acids.
Adverse effects and other considerations
- all NSAIDs carry increased cardiovascular risks (naproxen has replaced diclofenac in the UK and US after such concerns; more recently similar concerns have been raised over long-term use of ibuprofen), increase in blood pressure and reduced anti-aggregatory and vasodilation effects, increased risk of bleeding (especially gastrointestinal (GI) tract, aggravated by accumulation of these acidic chemicals in the GI mucosal cells by ion trapping when orally administered);
- protection of the GI tract under NSAID treatment by prescribing additional antacid medications may be advised;
- diclofenac is known to be a cause of common intolerance and allergic reactions, causing mainly urticaria (rash and swelling) and other skin problems, but anaphylaxis (allergic shock) has been reported;
- many NSAIDs are available over the counter but only in low-dose preparations; these may be inadequate for pain management in some situations.
Typical maxillofacial applications of NSAIDs include treatment of
- acute bone pain after surgery or after trauma such as fractures of facial bones (it may be better to avoid NSAIDs because there is some evidence that these agents may delay wound healing);
- acute orofacial infections;
- in combination with other analgesic agents such as paracetamol (see below) or opioids (see below) in the treatment of more severe pain (see above, Table 1).
Opioids
Mechanism
Opioids all act upon the same group of receptors, known as opioid receptors and belonging to a family of G protein-coupled receptors (GPCRs). GPCRs are part of a large family of proteins that reach across the cell membrane (transmembrane domains) and are involved in numerous regulatory processes in health and disease. Many different ligands bind to these receptors, including naturally occurring opioid-like substances released in the body (endogenous opioids such as β-endorphins and enkephalins) as well as synthetic opioids. Most opioid receptors are located in the central nervous system, but some are found in the periphery (peripheral opioid receptors). The binding of opioids to opioid receptors in the brain causes suppression of pain signals by descending pathways (descending inhibition). The descending pathways are mechanisms that modulate the pain signalling by fibres descending from the brain by interfering with the transmission of pain signals to the brain. There are inhibitory as well as enhancing mechanisms.
There are three main categories of opioid receptors, mu (µ), delta (δ) and kappa (κ). Their structures are generally similar, with the binding pocket of each being very similar but differing in the structures that make first contact with a ligand before it binds. This allows for degrees of selectivity for binding of ligands, depending on the ligand structures.
When opioids bind to the receptor(s), the receptors inhibit voltage-dependent calcium channels (Figure 3) or activate potassium channels. This action stops the pain signal from travelling along the neuron and is the main mechanism of analgesia by opioids.
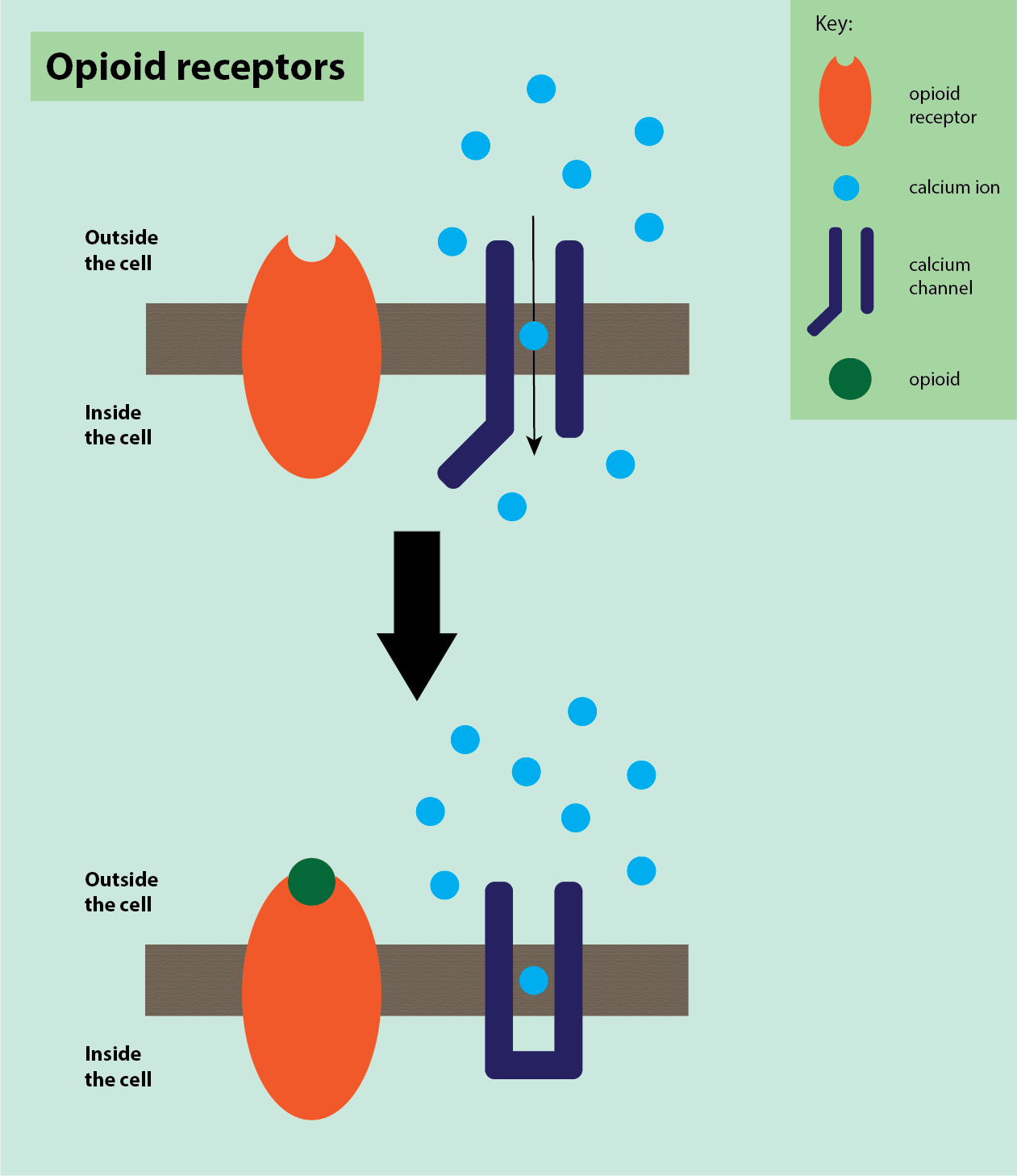
The effect of binding depends on whether the receptor is on the pre-synaptic or post-synaptic neuron. Binding to opioid receptors on the pre-synaptic neuron inhibits voltage-dependent calcium channels, so decreasing the release of neurotransmitters such as glutamate, serotonin, acetylcholine and substance P. Binding to opioid receptors on the post-synaptic neuron activates potassium channels, which stop depolarisation, inhibiting neuronal firing by decreasing neuronal excitability and causing hyperpolarisation of the neuron.
Opioids can also interact with α2-adrenergic receptors on pre-synaptic neurons, which are linked to inhibition of norepinephrine (an excitatory neurotransmitter) release, and cause a decrease in norepinephrine, so inhibiting neuronal excitability and firing by yet another mechanism.
In general, opioids overall have a significant inhibitory effect on physiological pain sensation, but cause different analgesia and tolerance in different conditions. The additional effects of opioids on mood clearly have a higher-function impact on the perception of the sensation known as pain.
List of opioids commonly used in maxillofacial surgery
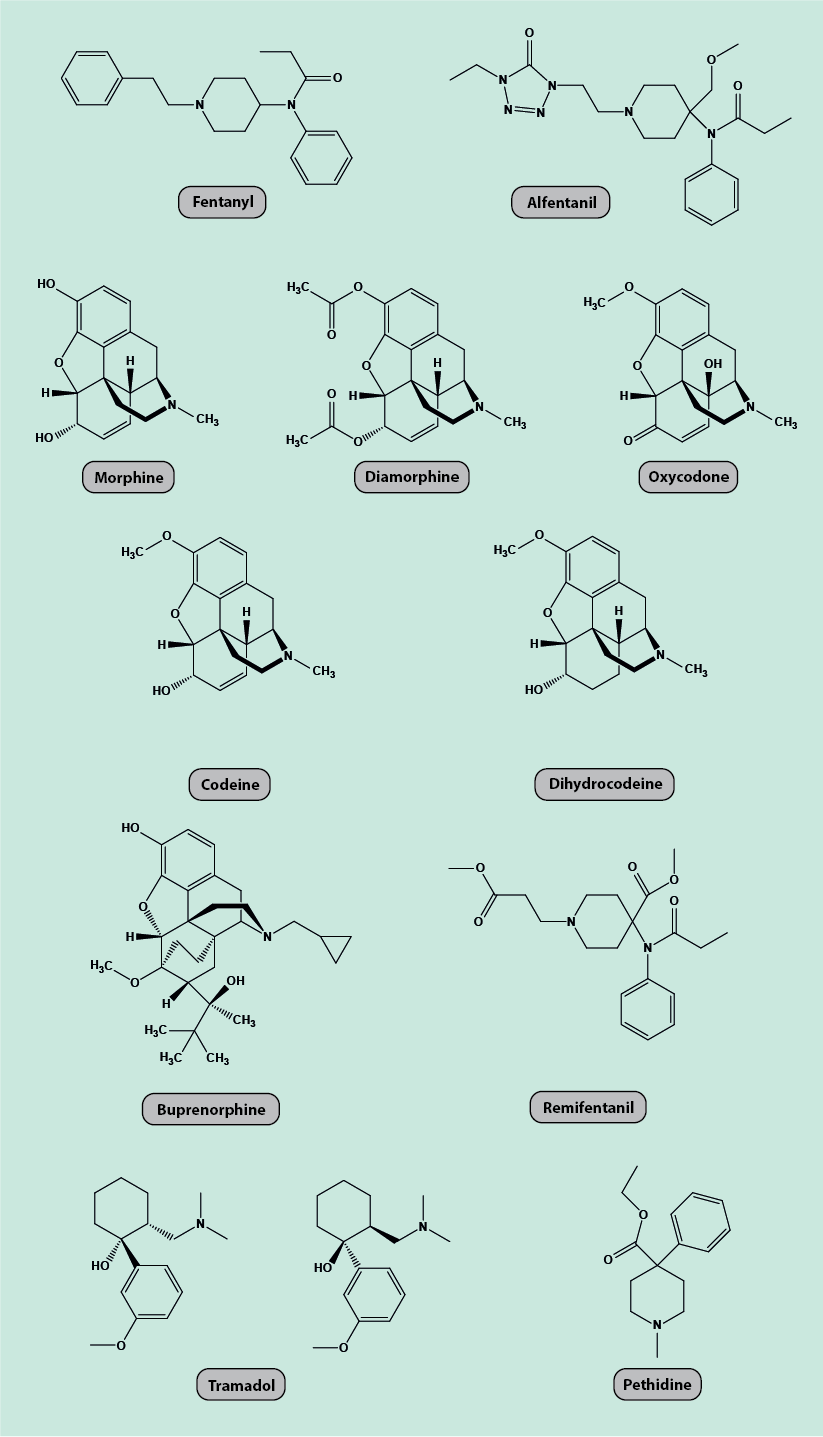
Figure 4 depicts the chemical molecular structures of eleven commonly used opioids (fentanyl, alfentanil, remifentanil, morphine, diamorphine, oxycodone, pethidine, buprenorphine, tramadol, codeine, dihydrocodeine). Some agents fall into groups of chemically closely related substances.
Adverse effects and other considerations
- sustained opioid consumption can produce paradoxical pain – nociceptive sensitisation (lowers pain thresholds) and analgesic tolerance;
- respiratory effects, cough suppression, nausea, vomiting, constipation and dependence (tolerance and addiction);
- codeine is a pro-drug that first needs to be metabolised in the body to its active form (morphine). There is a wide variation of genetic patterns in the population with regard to the level of codeine metabolism which makes dosage difficult. For some people codeine does not work because it is not sufficiently converted, whereas for others the conversion rate is very high, potentially leading to unsafe high morphine levels and related adverse effects (a particular concern in children and frail elderly people, regarding respiratory failure);
- a number of opioids can be delivered in non-oral ways that are helpful for maxillofacial patients when swallowing of pills is a problem. For example, fentanyl can be delivered by a skin patch, buprenorphine is available in a preparation for sublingual absorption through the oral mucosa.
Typical maxillofacial applications of opioids include
- role as part of general anaesthesia medications;
- role as part of heavy sedation, for example in intensive care;
- pain control in late stages of palliation of cancer pain.
Sedatives
Mechanism
There are different mechanisms of action for different classes of sedatives and these mechanisms of action are still incompletely understood. The general effect of sedatives appears to be to depress spontaneous and evoked activity of neurons, especially synaptic transmission. Three examples of different classes of sedatives are (there are more classes than these three)
- GABAA receptor agonists;
- non-competitive NMDA antagonists;
- α2-adrenoceptor agonists.
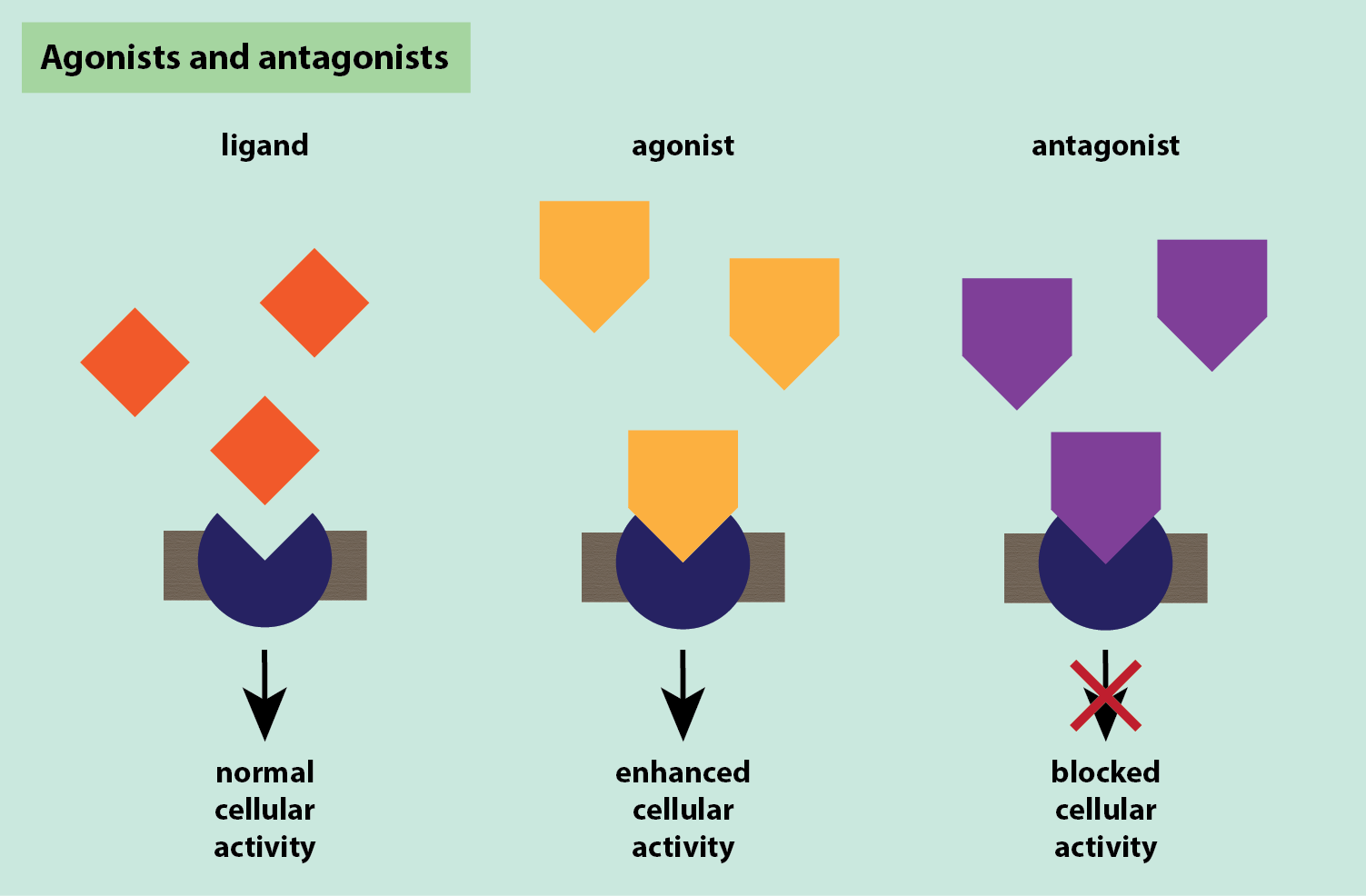
Agonists mimic an effect and activate their target, often enhancing its activity, whilst antagonists block the usual action of their target (Figure 5).
GABAA receptor agonists bind to an allosteric site (a place on the receptor where a molecule that is not the usual substrate can bind) on the GABAA receptor (Figure 6).
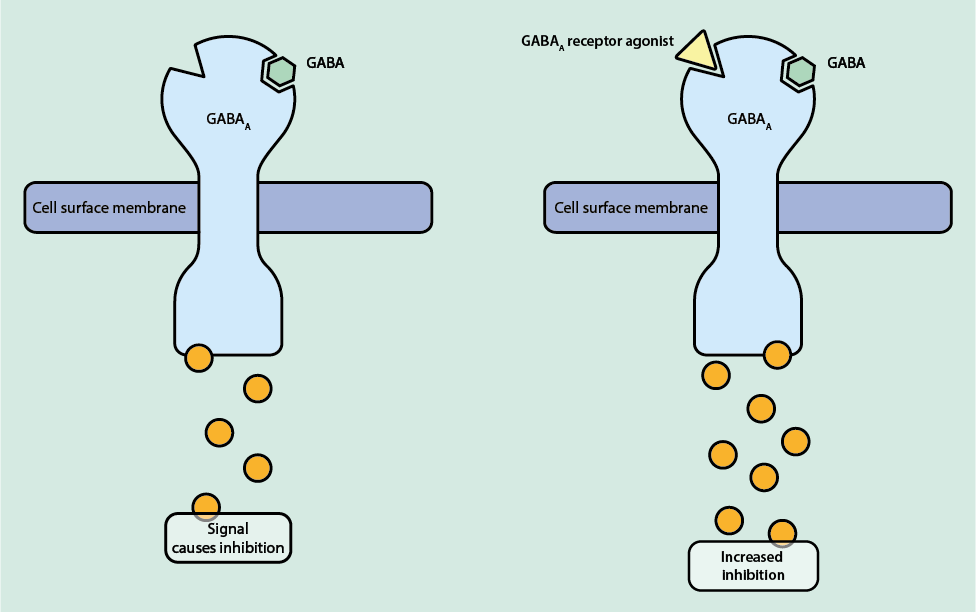
The binding of GABA to its receptor causes a conformational change, opening the channel and allowing Cl- ions into the cell. This causes increased negative charge inside the cell, causing hyperpolarisation of the neuron and decreasing the chance of an action potential (inhibition). The agonists do not activate the GABAA receptor directly but enhance the effect of GABA, making an action potential even less likely and so intensifying the inhibitory effect.
Non-competitive NMDA antagonists inhibit the action of the NMDA receptor (N-methyl-D-aspartate receptor, an ion channel in nerve cells) by binding to an allosteric site on the receptor and deactivating it. The NMDA receptor is a glutamate (an excitatory neurotransmitter) receptor. When glutamate binds to the NMDA receptor, it causes ion channels to open, allowing Ca2+ ions into the cell, making the inside of the cell more positive, causing the formation of an action potential and a pain signal to be felt. Non-competitive NMDA antagonists bind to the NMDA receptor so that, when glutamate binds, the channels do not open, so stopping the formation of an action potential and inhibiting the pain signal.
The mechanism of action of α2-adrenoceptor agonists for pain inhibition is currently poorly understood. These agents are thought to bind in an allosteric site and activate α2-adrenergic receptors, which are inhibitory in nature. Activation of α2-adrenergic receptors on post-synaptic neurons then causes increased outwards conductance of potassium ions (K+), reducing neuronal excitability and so inhibiting neuronal firing and stopping a pain signal from being felt. Many α2-adrenergic receptors on pre-synaptic neurons are linked to inhibition of norepinephrine (an excitatory neurotransmitter) release. It is suggested that α2-adrenoceptor agonists may enhance the effect of α2-adrenergic receptors on pre-synaptic neurons, causing a decrease in neurotransmitter signalling and so inhibiting the pain signal.
List of sedatives commonly used in maxillofacial surgery
Figure 7 depicts the chemical molecular structures of five commonly used sedatives (diazepam, midazolam, propofol, ketamine, clonidine). Diazepam, midazolam and propofol act as GABAA receptor agonists, ketamine is a non-competitive NMDA antagonist, and clonidine is a α2-adrenoceptor agonist. Ketamine and clonidine are often administered together as co-analgesics.
Adverse effects and other considerations
- ketamine can cause psychoses and/or hallucinations, increased blood pressure (hypertension);
- propofol can cause hypotension (low blood pressure) and convulsions;
- clonidine can cause hypotension and bradycardia (fast heart beat).
Typical maxillofacial applications of sedatives include
- intravenous application to augment local anaesthesia for surgery;
- propofol (in the UK) used as an induction agent in general anaesthesia;
- clonidine is mainly used in intensive care settings.
It is worth noting that the definitions of intravenous sedation and intravenous anaesthesia differ quite radically in different countries. UK practice and North American practice are totally different in relation to the drugs used and who administers them. The UK limits the surgeon to single-agent techniques whereas the US routinely uses combinations delivered by a surgeon in an ‘office’ environment.
Local analgesics
Mechanism
Local analgesics act by reversibly blocking impulse transmission in peripheral nerves, blocking a pain signal from being felt (Figure 8).
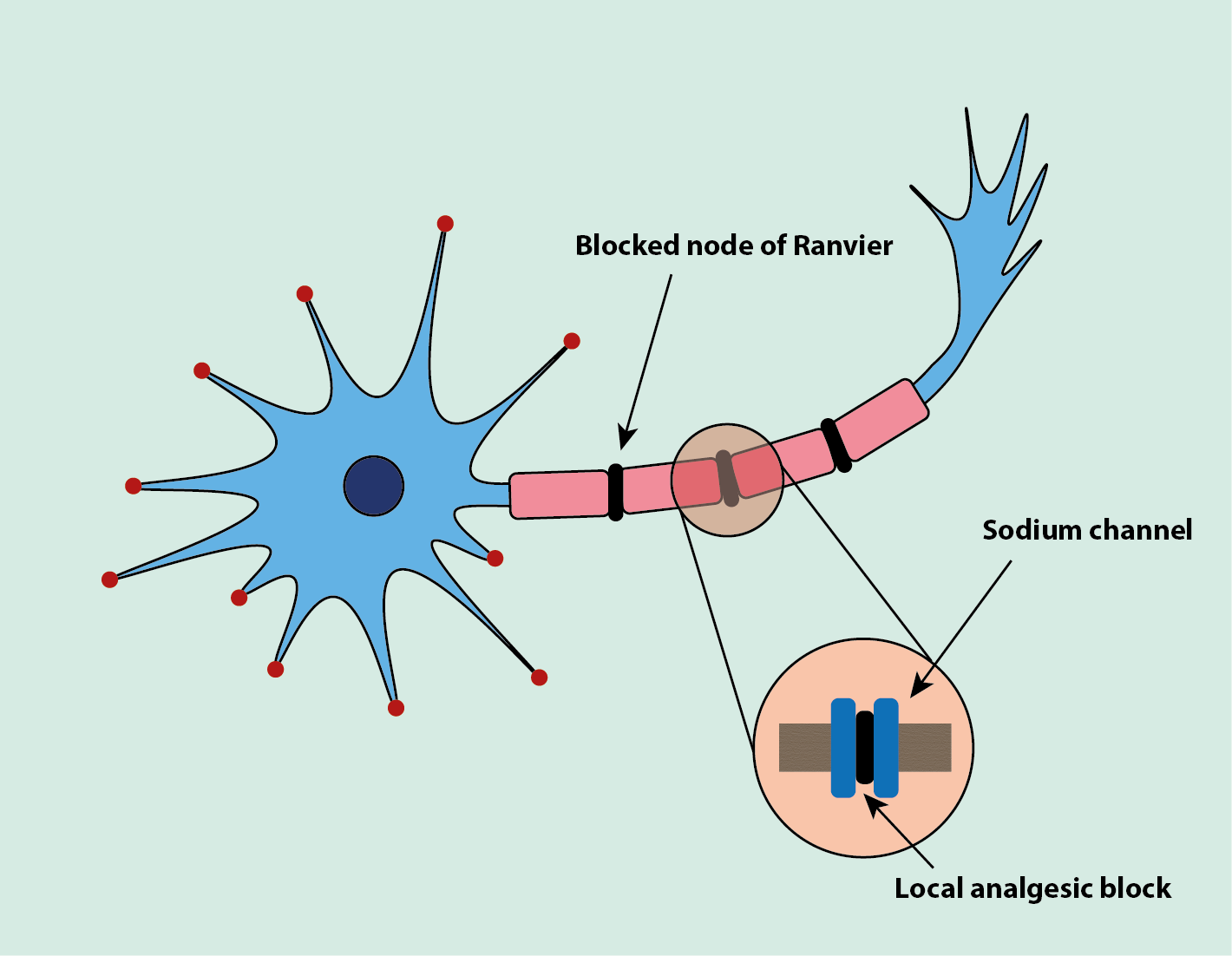
Local analgesics in their soluble form are injected as solutions in water. In tissues, a proportion of the drug dissociates to a lipid and a soluble free base, which can cross the neuronal lipid membrane. The free base blocks the sodium channels on the neuron which blocks the nerve action potential (Figure 8). The small myelinated neurons are blocked first, with larger unmyelinated neurons being blocked last. The temporary loss of functions caused by the blocking occurs in the order: pain sensation => temperature sensation => sensitivity to mechanical stimulus => motor functions.
List of local analgesics commonly used in maxillofacial surgery
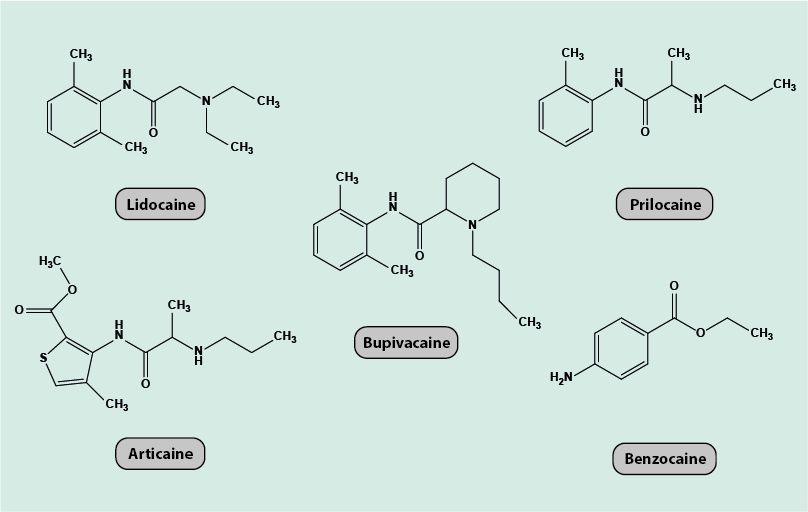
Figure 9 depicts the chemical molecular structures of five commonly used local analgesics (lidocaine, prilocaine, articaine, benzocaine, bupivacaine). These agents share a common molecular motif of an aromatic (usually 6-membered) ring structure joined to an amine group.
Adverse effects and other considerations
- allergic reactions;
- accidental injection directly into the blood stream;
- lidocaine does not select between Na+ ion channels in neurons and those in other fibres;
- intra-nerve bundle (for example, inferior alveolar nerve) injection has been associated with permanent nerve damage.
Typical maxillofacial applications of local analgesics include
- many intraoral and dentoalveolar procedures are performed under local anaesthesia in an outpatient setting;
- as local anaesthesia peri-operatively;
- postoperative infiltration of surgical wounds with local anaesthetics (including donor sites in reconstructive surgery);
- sometimes injected prior to incision in surgical procedures under general anaesthesia both as haemostatic agent (in adrenaline-containing preparations) and as pre-emptive analgesic or to reduce the amount of narcotic anaesthetic used in general anaesthesia;
- some preparations of local analgesics (benzocaine in particular, although it is associated with a higher incidence of hypersensitivity reactions) can be applied topically (gels or mouthwash) and are used to ease eating and/or oral hygiene postoperatively, or with painful mucositis from radiotherapy or chemotherapy, or other painful oral ulcerations and conditions.
Cyclooxygenase-2 (COX-2) inhibitors
Mechanism
Cyclooxygenase-2 (COX-2) inhibitors, like non-steroidal anti-inflammatory drugs (NSAIDs; see above), have analgesic, antipyretic (prevent or reduce fever) and anti-inflammatory effects. Their mechanism of action is very similar to that of NSAIDs, with the main difference being that NSAIDs block COX-1 and COX-2 enzymes, whereas COX-2 inhibitors only block the COX-2 enzymes and thus the synthesis only of those prostaglandins involved in response to tissue damage, causing inflammation and pain (Figure 10). This degree of selectivity holds promise to cause fewer adverse effects (on the GI tract) than traditional NSAIDs.
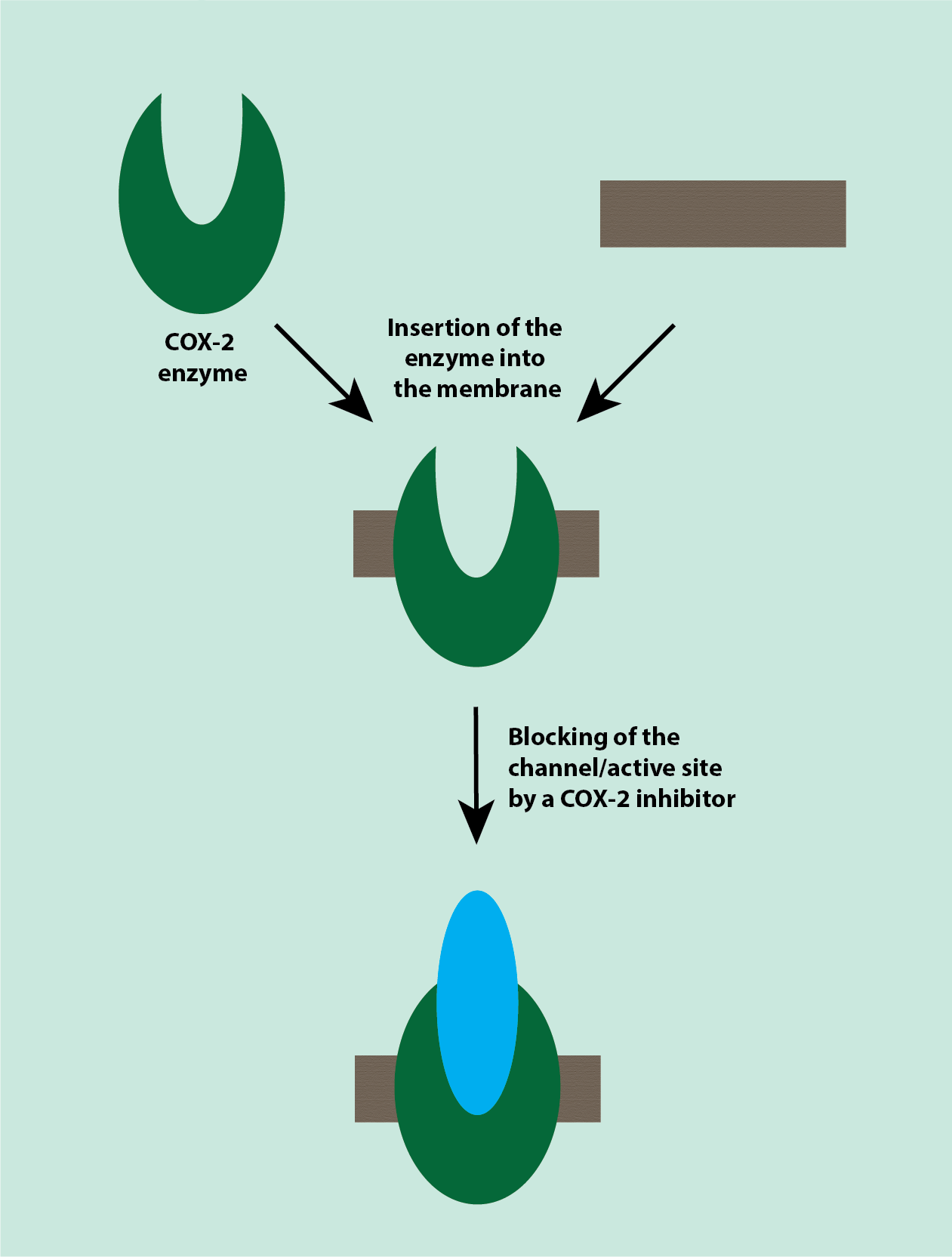
There is a side pocket in the catalytic site of the COX-2 enzyme, which is not present in the COX-1 enzyme. COX-2 inhibitors use this side pocket to selectively inhibit the COX-2 enzyme and not the COX-1 enzyme.
COX-2 inhibitors are thought to decrease endocannabinoid metabolism by inhibiting the COX-2 enzyme, which metabolises endocannabinoids. This causes an increase in endocannabinoids (naturally occurring substances in the body’s metabolism; chemically closely related to some of the components in cannabis (see below)), which have an analgesic effect. COX-2 inhibitors are less potent than non-steroidal anti-inflammatory drugs.
The better selectivity of COX-2 inhibitors compared with traditional NSAIDs would appear to make COX-2 inhibitors attractive anti-inflammatory analgesic agents. Unfortunately, COX-2 inhibitors significantly increase the risk of cardiovascular complications and, therefore, are not first-line anti-inflammatory analgesic agents.
Examples of COX-2 inhibitors
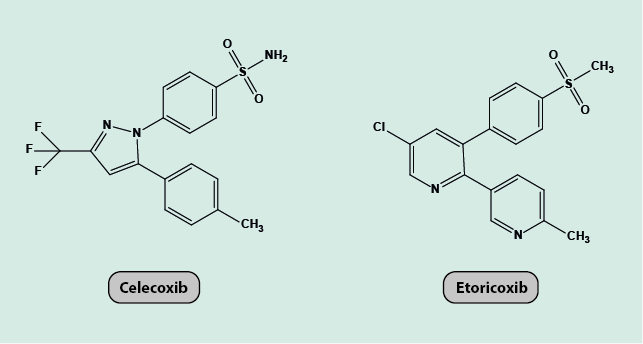
The molecular chemical structures of two examples of COX-2 inhibitors (celecoxib, etoricoxib) are depicted in Figure 11.
Adverse effects and other considerations
- hypertension and reduced diuresis (water retention);
- significantly increased risk of myocardial infarction and stroke;
- no impairment of platelet aggregation;
- potentially reduced adverse effects on GI tract in comparison with NSAIDs.
Typical maxillofacial applications of COX-2 inhibitors include
- essentially not used in oral and maxillofacial surgery;
- possible role in degenerative arthritic conditions of the temporomandibular joint (jaw joint) but no definite data and results yet.
Other common drugs
Paracetamol
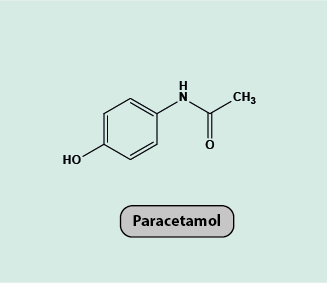
Paracetamol (Figure 12) is a non-acidic antipyretic (prevent or reduce fever) analgesic. It is not known precisely where in the pain signalling cascade paracetamol works. It has been suggested that it is a relatively weak inhibitor of cyclooxygenase (COX) enzymes in the brain, inhibiting prostaglandin synthesis, but is not active in peripheral cyclooxygenase sites. The high lipophilicity (high solubility in fatty tissue) and the non-acidic nature of paracetamol are thought to facilitate permeation of paracetamol across the blood-brain barrier and into the central nervous system.
Paracetamol lacks any significant anti-inflammatory properties. It has been suggested that this is due to its non-acidic nature and the resulting absence of enrichment in inflamed tissue, which is acidic. The analgesic effects of paracetamol tend to be weak due to low efficacy and so paracetamol is typically only suitable to treat mild pain (yet many anaesthetists swear by its efficacy when administered intravenously peri- and post-operatively).
Similar to the COX-2 inhibitors (see above), paracetamol is thought to decrease endocannabinoid metabolism by inhibiting the COX-2 enzyme. This causes an increased concentration of endocannabinoids, which have an analgesic effect.
Paracetamol causes increased blood pressure, an increased risk of cardiovascular events and is liver toxic (due to the production of reactive intermediates) but lacks the gastrointestinal tract toxicity and acute renal (kidney) toxicity that non-steroidal anti-inflammatory drugs have. Paracetamol is an example of a drug with a narrow therapeutic window, with only a small difference in dose required for analgesic effects and doses that lead to liver damage. Paracetamol is particularly popular in the UK.
Paracetamol is the standard first line analgesic for most people with toothache and is responsible for multiple cases of overdose related liver damage, including death. It is also used extensively in combination with codeine (see above) as a second line analgesic.
Metamizole
Metamizole (Figure 13), like paracetamol, is a non-acidic antipyretic (prevent or reduce fever) analgesic. It is thought to have a similar mechanism of action as paracetamol, as an inhibitor of cyclooxygenase (COX) enzymes in the brain. It has similar high lipophilicity and non-acidic properties as paracetamol, facilitating permeation across the blood-brain barrier and into the central nervous system. Metamizole has no significant anti-inflammatory properties.
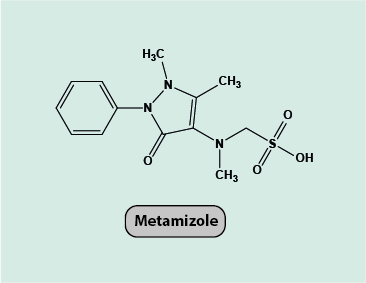
Metamizole lacks the gastrointestinal tract toxicity and acute renal toxicity that non-steroidal anti-inflammatory drugs have. However, metamizole does have blood toxicity and can cause rare but serious adverse effects (agranulocytosis).
Metamizole is not licenced in the UK but is widely used in Europe as a general analgesic as it is considerably more potent than paracetamol. It is an effective analgesic for bone pain and can be used on its own or in combination with other analgesic agents.
Botulinum toxin
Botulinum toxin is a highly poisonous substance that acts as a powerful paralytic neurotoxin. Botulinum toxin works by inhibiting the release of the ubiquitous neurotransmitter acetylcholine. Acetylcholine is an excitatory neurotransmitter that works at the neuromuscular junction. When acetylcholine is released by motor neurons, it activates the muscle. When acetylcholine is blocked, the muscle cannot be activated, so the muscle is paralysed (Figure 14).
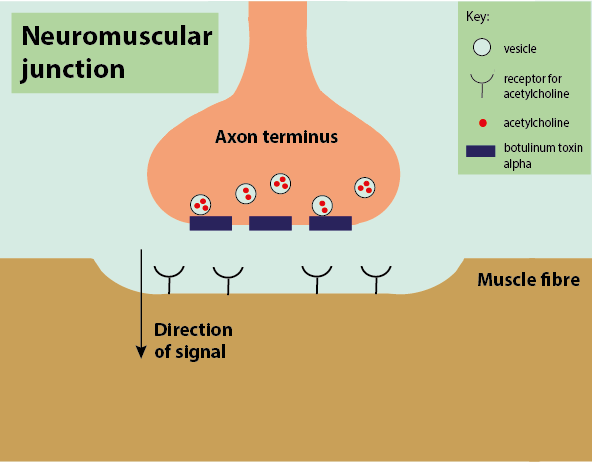
Botulinum toxin, amongst its many other medicinal applications, can act as an effective analgesic in treating painful muscle spasms by temporarily paralysing the muscle.
Botulinum toxin is extensively used to treat myofascial pain or facial arthromyalgia (painful dysfunction syndrome of the temporomandibular (jaw) joint) and is demonstrably effective for periods of several months, sometimes needing repeating injections.
Cannabinoids
Cannabinoids are natural (synthesised in the body, endocannabinoids) and synthetic compounds that interact with cannabinoid receptors. Cannabis is made up of many components of different cannabinoid ligands made up of many. Two such active components are tetrahydrocannabinol (THC) and cannabidiol (CBD) (Figure 15).
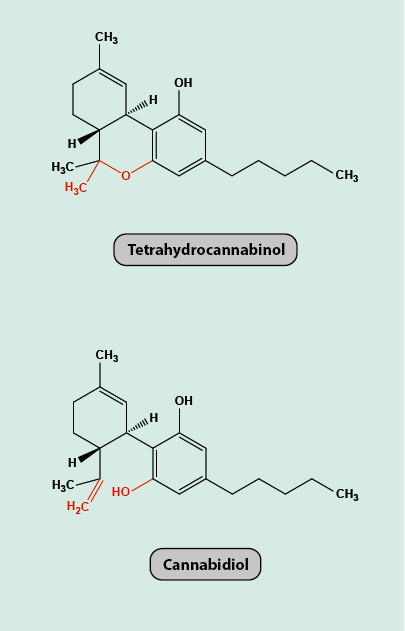
The two molecules have very similar structures but different effects. THC has relaxing psychoactive and/or analgesic effects, whereas CBD is not psychoactive but is suspected to hamper cognitive abilities. Depending on the preparation method, the ratios of THC and CBD vary. Natural cannabis products can have enhanced THC concentrations from selective breeding of the plants but this usually also enhances the CBD concentration. Medicinal cannabis preparations have controlled concentrations of THC extracts from plant material.
Cannabinoids act as analgesics in the brain, spinal cord and the peripheral nervous system. There are two main cannabinoid receptors, CB1 and CB2, although there are further receptors that cannabinoids can act on.
The mechanism of action of cannabinoids when bound to the cannabinoid receptors is not completely known. There are structural similarities between cannabinoid receptors and G protein-coupled receptors (opioid receptors). The two types of receptors and ligands are thought to have similar mechanisms of action. On pre-synaptic neurons, the binding is thought to cause inhibition of the voltage-dependent calcium channels (Figure 16), which causes a decrease in the release of neurotransmitters (such as substance P) and stops the pain signal from crossing the synapse.
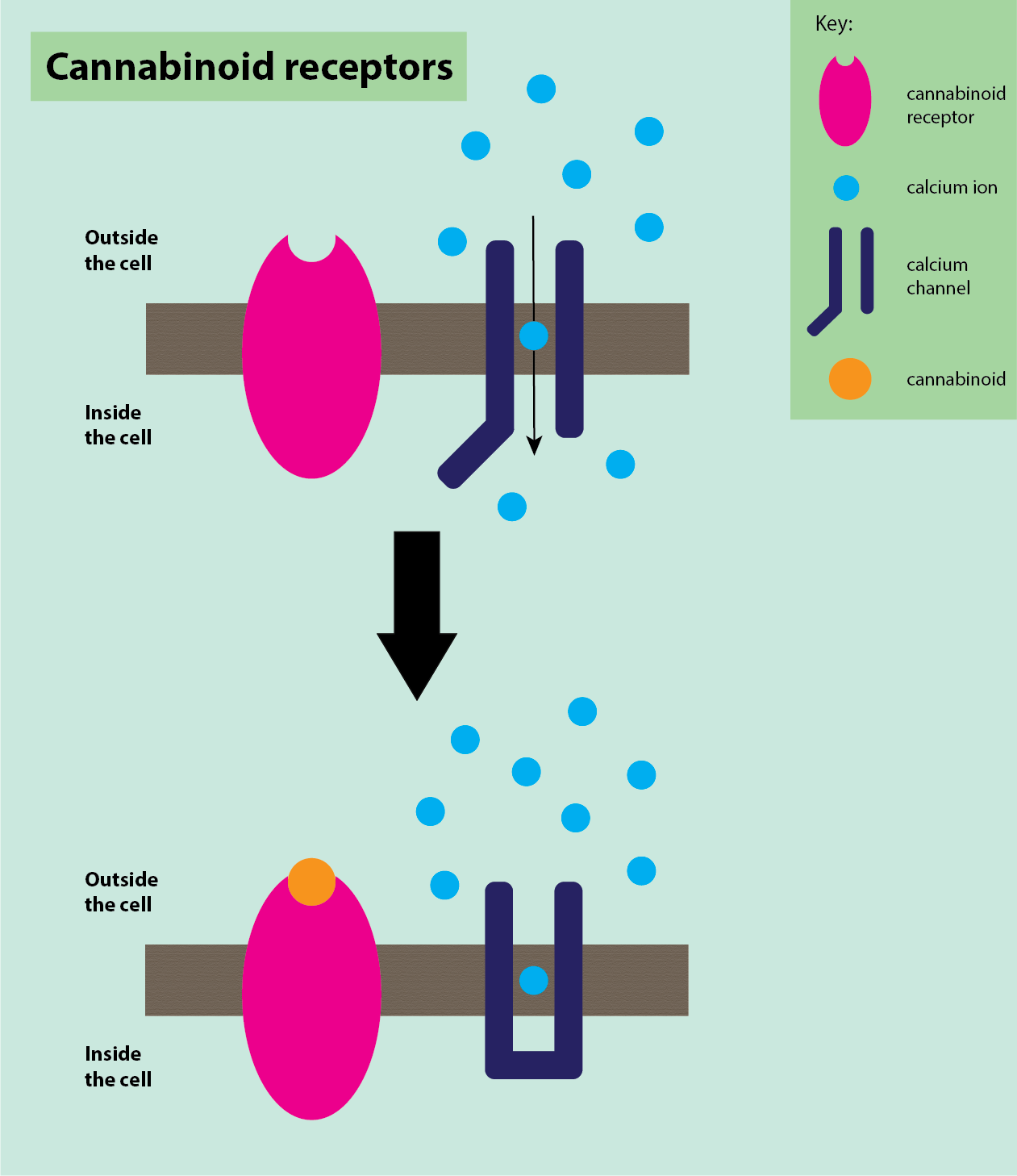
On post-synaptic neurons, binding of cannabinoids to cannabinoid receptors is thought to activate potassium channels, stopping depolarisation and inhibiting neuronal firing by decreasing neuronal excitability. Cannabinoids are also thought to cause some pain relief by descending inhibition, similar to opioids (see above).
Cannabinoids have an inhibitory effect on neuropathic pain, for example in neurodegenerative conditions such as multiple sclerosis or Parkinson’s disease, or can reduce / prevent seizures in some epilepsy conditions. The main analgesic effect of cannabinoids is thought to be a reduction of hyperalgesia and allodynia.
A preparation of medicinal THC and CBD extracts in the form of an oral spray has been used as part of palliation in oral squamous cell carcinoma and may actually have a direct cellular effect. There are certainly arguments for its systemic use as an antiemetic (preventing vomiting), mood enhancing, appetite promoting analgesic but its use is not yet established in the mainstream.
Anticonvulsants/Antiepileptics and Antidepressants
Antiepileptics/anticonvulsants and antidepressants both have a second life / fringe activity as analgesics. These agents are not designed as analgesics but have been found to act in some cases, in low dosage well below that needed for antiepileptic / antidepressant effect, as analgesics in the treatment of chronic and neuropathic pain conditions. The mechanisms of action of these agents as analgesics are poorly understood but are thought to work in a number of different locations.
Gabapentin and pregabalin
Gabapentin and pregabalin (Figure 17) are analogues of GABA. Both act as antiepileptics and neuromodulators. Despite being GABA analogues, they have no activity against GABA receptors, channels or transporters.
The mechanism of analgesic action of gabapentin and pregabalin is thought to be due to their high affinity for voltage-gated calcium channels, causing a reduction in the influx of calcium ions into cells and thus stopping the release of excitatory neurotransmitters at synapses in the spinal cord.
Pregabalin is more potent than gabapentin, so is more likely than gabapentin to have dose levels that are both effective and tolerable in the treatment of neuropathic pain.
Gabapentin and pregabalin are regularly used for neuropathic pain of a variety of origins. The two drugs are less effective in treating trigeminal neuralgia.
Carbamazepine
Carbamazepine (Figure 18) is also an antiepileptic and is the drug of choice for treatment of trigeminal neuralgia. It is also used in the diagnosis of this condition because of its unique efficacy for this condition.
The main mechanism of action of carbamazepine is by binding to inactivated sodium channels and stopping them from becoming active again, effectively blocking voltage-gated sodium channels and stopping the action potential moving along the neuron. Carbamazepine also has some action against voltage-gated calcium channels, similar to the working mechanisms of gabapentin and pregabalin (see above), and it increases the amount of the inhibitory neurotransmitter GABA.
Fluoxetine and Phenothiazine
Fluoxetine (Figure 19) is an antidepressant agent with a slightly stimulating effect.
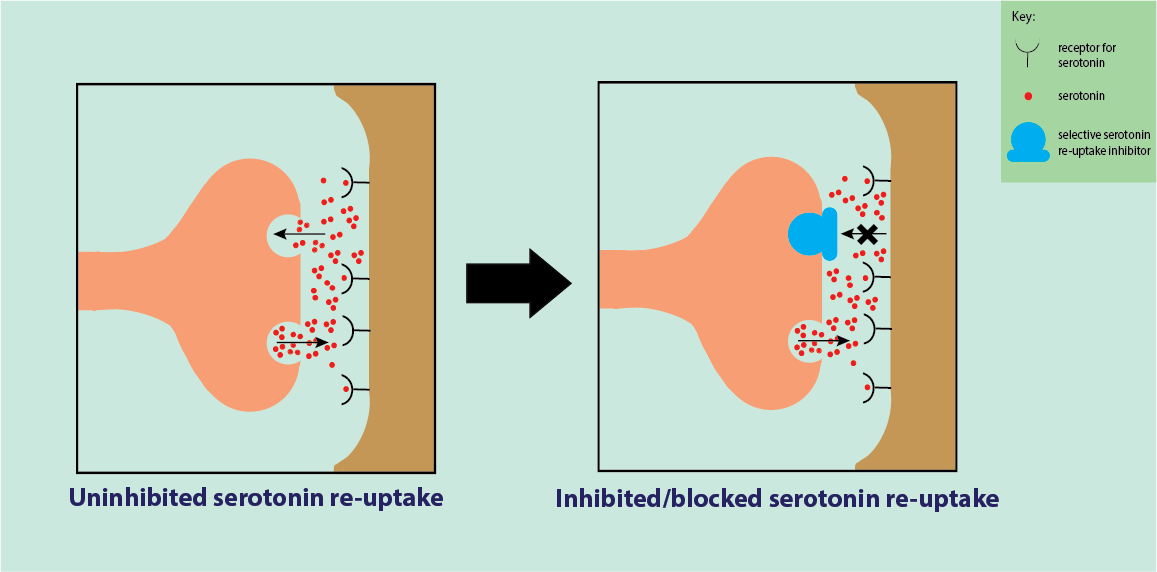
It acts as a selective serotonin re-uptake inhibitor, blocking serotonin (a neurotransmitter) re-uptake in synapses in the central nervous system. This allows serotonin to stay in the synapse for longer and have a greater prolonged inhibitory effect on the post-synaptic receptor, which can block pain signals (Figure 20). Selective serotonin re-uptake inhibitors act on multiple receptors with multiple effects.
Fluoxetine is sometimes combined with phenothiazine (Figure 19), a neuroleptic agent (an antipsychotic drug with a slight sedative action). This combination has been shown to be sometimes more powerful as an analgesic than an antidepressant drug alone.
Antidepressant agents have been shown to provide good pain relief in particular for allodynia (pain response from stimuli which do not normally provoke a pain sensation). Long-term use of antidepressants does carry a small but significant risk of gastrointestinal or post-surgical bleeding, but this is not a serious risk when used in low dose for the treatment of neuropathic pain conditions.
Amitriptyline and Nortriptyline
Amitriptyline and nortriptyline are tricyclic antidepressant agents with potential for neuropathic pain relief (Figure 21).
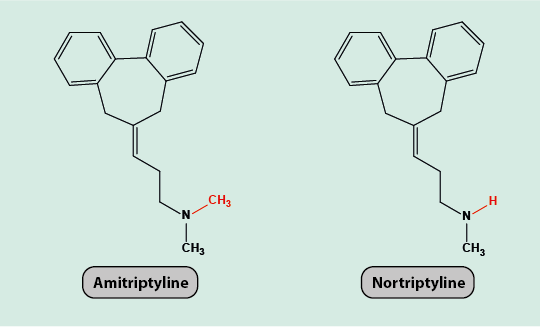
Amitriptyline interacts with many different receptors on nerve cells, including opioid and histamine receptor sites. It is also known to have some blocking effects on sodium and potassium ion channels in the central nervous system as well as reducing the re-uptake of noradrenaline. All of these mechanisms are thought to contribute to the analgesic effects of amitriptyline (and other tricyclic antidepressants) as they all play a role in chronic neuropathic pain conditions.
Amitriptyline is a pro-drug and is metabolised to nortriptyline (Figure 21). Nortriptyline hinders the re-uptake of both serotonin and noradrenaline (neurotransmitters) at different types of neurons. This gives a sedative effect at low doses and can improve sleep when taken at night.
Antidepressants have been shown to provide good pain relief in particular for allodynia (pain response from stimuli which do not normally provoke a pain sensation). Long-term use of antidepressants does carry a small but significant risk of gastrointestinal or post-surgical bleeding, but this is not a serious risk when used in low dose for the treatment of neuropathic pain conditions.
Miscellaneous treatments
As far as management of pain, in particular chronic pain, is concerned, there are a number of non-invasive options with poor evidence of efficacy – but which at least are likely to do little harm (other than to the wallet). Such non-invasive options include
- cognitive behavioural therapy and other forms of talking therapies;
- relaxation techniques such as biofeedback and similar approaches;
- acupuncture;
- application of low-intensity (low-level) laser (or other light) therapy, advertised in alternative medicine;
- transcutaneous electrical nerve stimulation (TENS) is claimed to provide short-term pain relief in some instances.
If conventional (and alternative) medical therapies fail to relieve pain, more invasive techniques such as cryotherapy, peripheral nerve blocks, or percutaneous destruction of a nerve ganglion (commonly the trigeminal nerve) may be necessary. Sometimes neurosurgical interventions such as microvascular decompression of the root of the trigeminal nerve are indicated where it has been demonstrated that a microvascular loop compresses the trigeminal nerve. Overall, invasive methods for the management of pain conditions are rarely indicated.
There are numerous areas of overlap between medical management of pain, as discussed in this section, and working principles of general and local anaesthesia as well as intensive care.